Examples of Single Gene Disorders
Adenosine deaminase (ADA) deficiency
What is ADA deficiency?
ADA deficiency is one form of SCID (severe combined immunodeficiency, see below). The immune system does not work properly, so the body is open to infection from bacteria and viruses.
Affected gene
The affected gene in ADA deficiency is ADA, on chromosome 20. The gene codes for ADA (adenosine deaminase) protein. People with the disorder have two non-working copies of the gene, and so they make no working ADA protein.
The job of ADA protein is to break down a toxic substance called deoxyadenosine, which forms naturally when DNA is broken down. Nearly all types of cells in the body make ADA protein. But it is especially active in cells of the immune system called B and T lymphocytes. Without ADA enzyme, the toxin builds up and destroys these infection-fighting immune cells.
ADA deficiency follows an autosomal recessive inheritance pattern. An affected child must inherit two non-working copies of the gene, one from each parent.
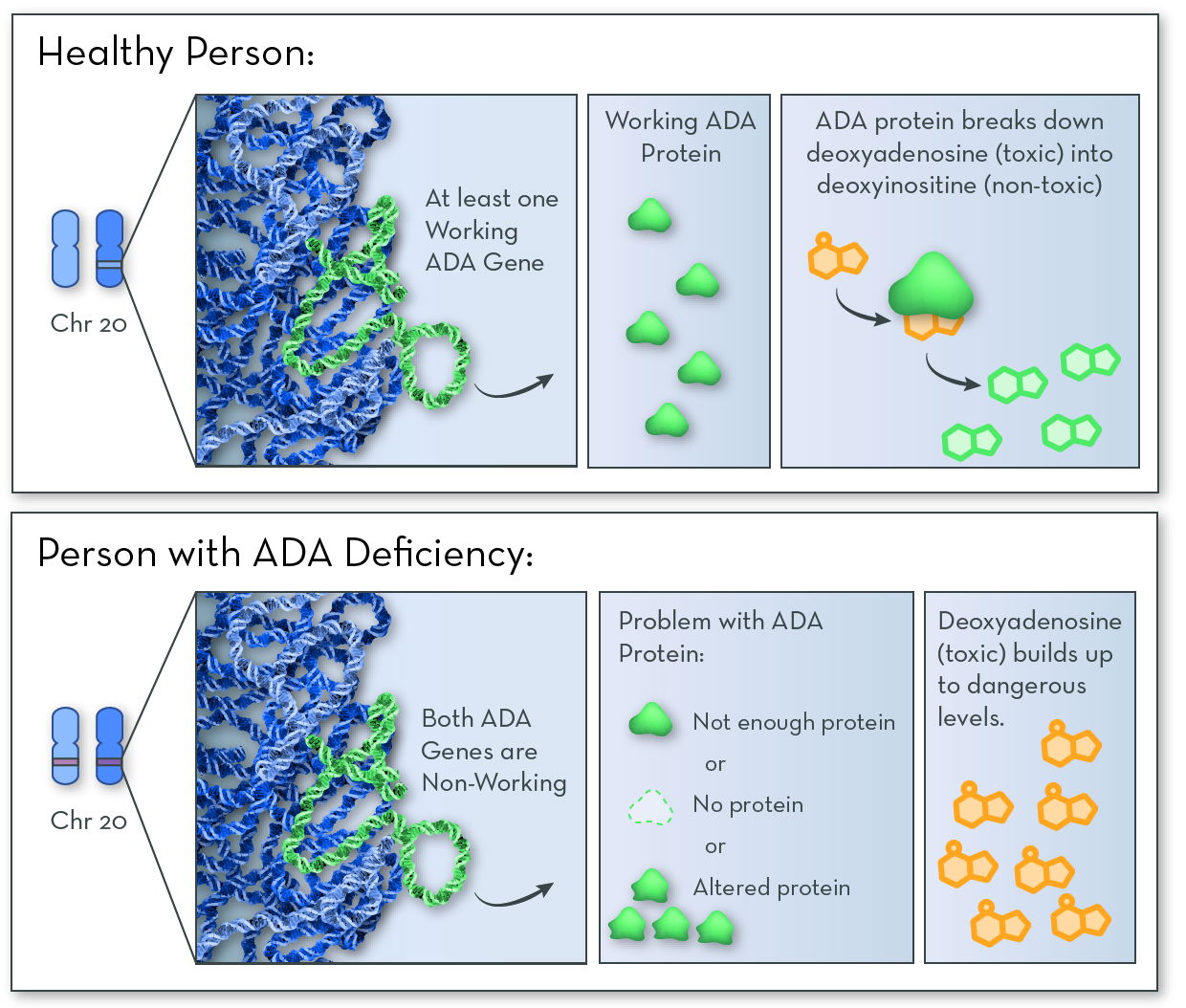
Without working ADA protein, the cell cannot break down a toxic substance called deoxyadenosine. The toxin builds up in infection-fighting immune cells, killing the cells and leaving the body open to infections.
Symptoms and features of ADA deficiency
ADA deficiency affects the immune system, leaving the body open to all kinds of infections. Infections of the skin, respiratory system, and digestive tract are common. Bacteria that are normally harmless can make someone with ADA deficiency very sick.
With no intervention, most babies who are born with the disorder die within a few months.
Treating and managing ADA deficiency
The goals with treating and managing ADA deficiency are to (1) prevent infection, and (2) provide a source of ADA protein so that immune cells can survive.
Ways to increase ADA levels:
- Bone marrow transplantation from a biological match (for example, a sibling) to provide healthy immune cells
- Transfusions of red blood cells (containing high levels of ADA) from a healthy donor
- Enzyme replacement therapy, involving repeated injections of the ADA enzyme
- Gene therapy - to insert synthetic DNA containing a normal ADA gene into immune cells
Interesting facts about ADA Deficiency
ADA deficiency is extremely rare. Only about 10 to 20 children are diagnosed with the disease in the United States each year.
In 1990, doctors William French Anderson, Michael Biase, and Ken Culver performed the first successful gene therapy on Ashanti de Silva, a young girl with ADA deficiency.
Alpha-1 Antitrypsin Deficiency
What is alpha-1 antitrypsin deficiency (Alpha-1)?
Alpha-1 affects the lungs and sometimes the liver. Alpha-1 is often misdiagnosed because its symptoms look like those of asthma, bronchitis, or smoking-induced emphysema. Many are not diagnosed until middle adulthood, and up to 95% are never diagnosed.
Affected gene
The affected gene in Alpha-1 is SERPINA1, on chromosome 14. This gene codes for a protein called alpha-1 antitrypsin. People with the disorder have two non-working copies of the gene, so they make no working alpha-1 antitrypsin protein.
Alpha-1 antitrypsin protein is normally made in the liver and released into the blood. It protects the lungs from attack by an enzyme called neutrophil elastase. Neutrophil elastase is made naturally by white blood cells (specifically neutrophils) in response to an infection or irritants. It helps to clear away damaged tissue in the lungs.
Without alpha-1 antitrypsin protein, neutrophil elastase attacks and damages the lungs. Some people who have Alpha-1 make no alpha-1 antitrypsin protein at all, and they suffer only lung damage. Others make a version of the protein that gets stuck in the liver. They suffer damage to both the lungs and the liver.
Alpha-1 follows an autosomal recessive inheritance pattern. An affected child must inherit two non-working copies of the gene, one from each parent.
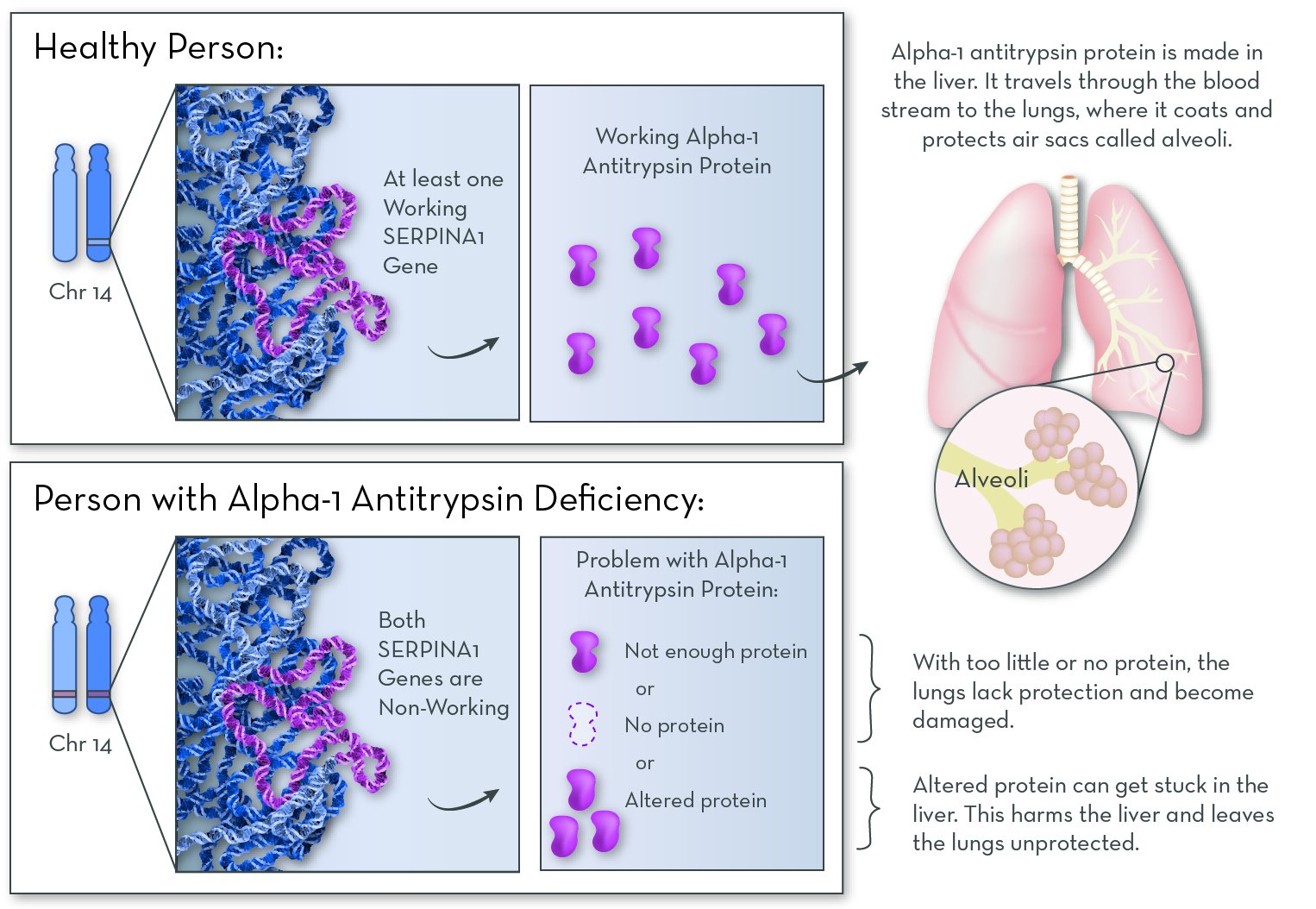
Alpha-1 antitrypsin protein is active in the lungs, where it protects them from damage. Without the protein, the lungs slowly accumulate damage over the course of a lifetime.
Symptoms and features of Alpha-1
The effects of Alpha-1 vary from person to person. The effects depend in part on the specific gene variants that a person has.
The main feature of Alpha-1 is damage to the tiny air sacs (alveoli) in the lungs. When the alveoli are damaged, the lungs cannot expand and contract very well. This can cause shortness of breath, coughing, or wheezing. Over time, many people develop lung diseases, such as emphysema, asthma, or chronic bronchitis.
About 10% of babies and 15% of adults with the disorder also have liver damage, which can develop into the chronic disease cirrhosis. In rare cases, hard and painful lumps may form under their skin, called panniculitis.
Treating and managing Alpha-1
Protein therapy, in which alpha-1 antitrypsin protein is added to the blood, can slow lung damage. The protein is collected from the blood of healthy donors.
Alpha-1 is a potential target for gene therapy, in which a modified virus delivers a working copy of the SERAPINA1 gene into the patient's cells.
Asthma medications, such as inhaled steroids or bronchodilators, can help to ease lung symptoms. In later stages, if organs become heavily damaged, patients may receive an organ transplant.
People with alpha-1 should avoid tobacco. Smoking damages the lungs by increasing irritation (and therefore levels of neutrophil elastase) and by blocking the protective effects of alpha-1 antitrypsin.
Interesting facts about Alpha-1
Alpha-1 is most common among caucasians. It affect 1 in 2,500 caucasians, making it one of the most common genetic disorders in this group.
More information
Alpha-1 FoundationThe Alpha-1 Foundation Genetic Counseling Program provides confidential genetic counseling to the Alpha-1 community. It is also an expert resource for health care professionals. The toll-free phone number is 1-800-785-3177.
American Lung Association
American Liver Foundation
Cystic Fibrosis (CF)
What is cystic fibrosis
Cystic fibrosis affects multiple organs in the body, including the lungs and digestive system.
Affected gene
The affected gene in CF is CFTR (cystic fibrosis transmembrane conductance regulator), on chromosome 7. The gene codes for CFTR protein. People with CF have two non-working copies of the gene, and so they make no working CFTR protein.
The job of the CFTR protein is to help maintain salt balance by moving chloride ions (from sodium chloride, or salt) out of cells. This process is key for maintaining a thin layer of mucus inside the lungs, digestive tract, and other organs. Without CFTR protein, salt becomes unbalanced, and the mucus becomes thick and sticky. The most serious effects are the lung cells. Mucus clogs the airways, and increases the risk of infection.
Thick mucus also blocks ducts in the pancreas, keeping digestive enzymes from getting into the intestines. Without these enzymes, the intestines cannot break down food to extract all of the nutrients that are needed for a person to grow and thrive.
CF follows an autosomal recessive inheritance pattern. An affected child must inherit two non-working copies of the gene, one from each parent.
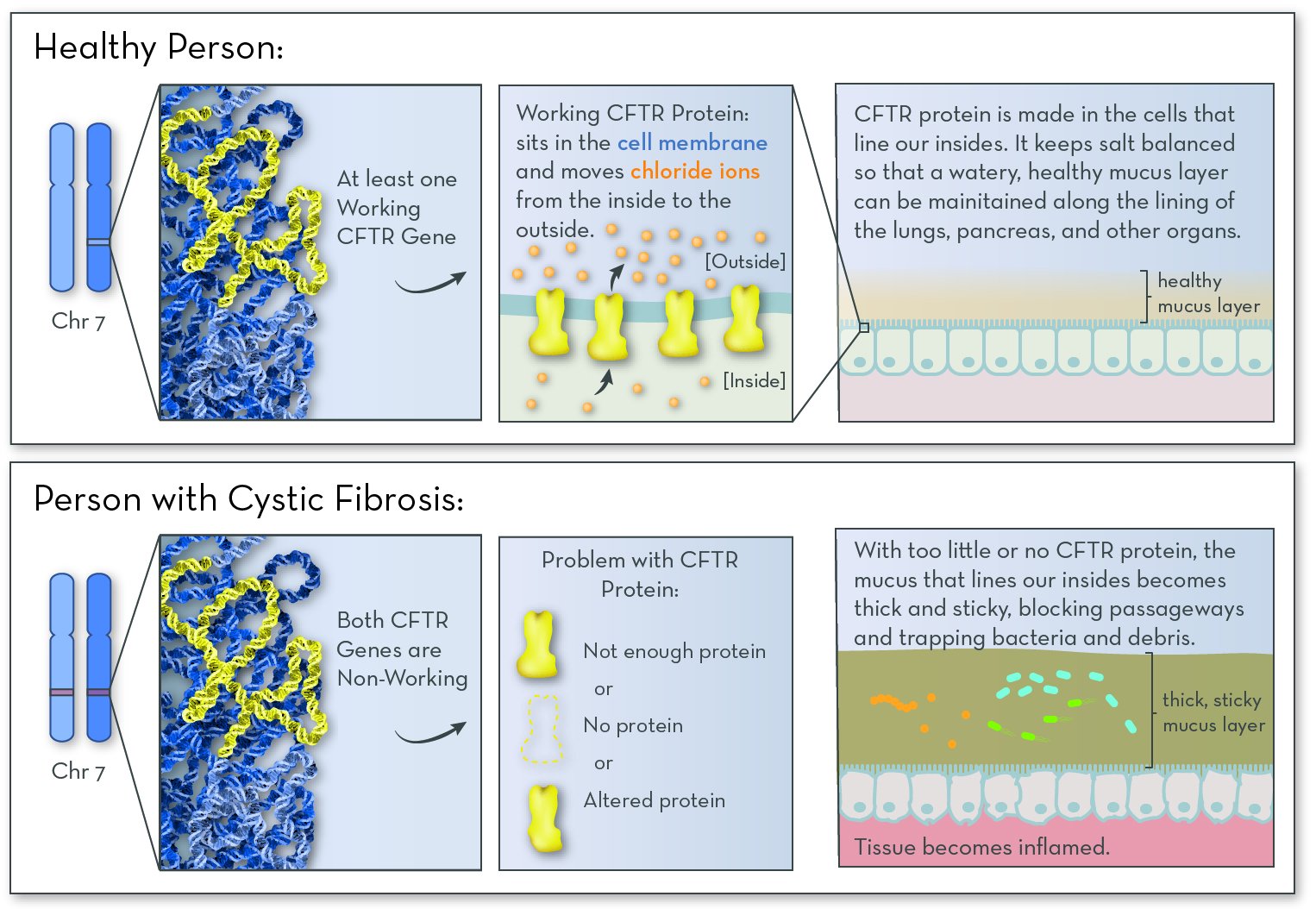
CFTR protein helps to maintain a healthy mucus layer along the insides of the lungs and pancreas. Without the protein, the mucus becomes thick and sticky. Mucus clogs the lungs and traps infection-causing bacteria. Digestive enzymes get stuck in the pancreas, causing problems with digestion.
Symptoms and features of CF
Babies with CF are often small, and all are prone to lung infections. Without treatment, most people with CF experience coughing or wheezing, respiratory illnesses (such as pneumonia or bronchitis), low weight, salty-tasting skin, and greasy stools. Challenges with salt regulation can lead to dehydration and salt imbalances.
The effects of CF vary, in part due to the specific disease-causing gene variants a person has. Some code for a CFTR protein with partial activity, and some produce no CFTR protein at all.
Early diagnosis leads to better outcomes. Because CF is fairly common, it is included in most newborn genetic screening panels. Starting treatment right away can prevent lung damage and improve nutrition, leading to a much longer and more fulfilling life.
Treating and managing CF
New treatments are helping people with CF live longer than ever. Lung therapies work by clearing mucus and preventing infections. Digestive enzyme pills taken with meals greatly improve nutrition, helping children grow and stay health.
- Chest therapy, in which a vibrating vest or repeated clapping on the back frees up mucus in the lungs
- Inhaled antibiotics to kill the bacteria that cause lung infections
- Bronchodilators (also used by people with asthma) to help keep the airways open
- Inhaled DNase, an enzyme that helps to thin sticky mucus
- Pancreatic enzyme replacement therapy to allow proper food digestion
CF is a potential target for gene therapy , in which a modified virus delivers a working copy of the CFTR gene into the patient's cells.
Interesting facts about cystic fibrosis
More than 1,000 different variations (alleles) of the CFTR gene have been identified in people with CF. The most common (in 70% of people with CF) is a three-base deletion in the DNA sequence, causing a single amino acid to be missing from the protein product.
Some evidence suggests that cystic fibrosis carriers are resistant to certain types of bacterial infections {remove link}
In the United States, about 2,500 babies each year are born with CF.
More than 10 million people in the US carry a CF-causing gene variation, but most don't know it.
Galactosemia
What is galactosemia?
People with galactosemia are unable to break down galactose, a type of sugar found in milk and other dairy products. Galactose is normally converted in the liver to the sugar glucose, which can provide energy for the cells of the body. But when it's not properly broken down, galactose is harmful to many of the body’s tissues.
Affected gene
The most commonly affected gene in galactosemia is GALT (galactose-1-phosphate uridylyltransferase). The gene codes for GALT protein. Most people with the disorder have two non-working copies of the GALT gene, and so they make no or very little working GALT protein.
The job of GALT protein is to help break down the sugar galactose.
Less common forms of galactosemia involve changes in different genes (GALK1 and GALE). These genes code for proteins that carry out different steps of galactose breakdown. These forms of galactosemia are usually less severe.
Most forms of galactosemia follow an autosomal recessive inheritance pattern. An affected child must inherit two non-working copies of the gene, one from each parent.

Treating and managing galactosemia
Galactosemia is managed through diet. Babies receive a galactose-free formula. Children with the disorder must stay away from foods and drinks containing galactose. These foods include milk, cheese, and legumes (dried beans).
Interesting facts
Galactosemia was first discovered in 1908 by the physician Von Ruess.
Classical galactosemia affects 1 in every 30,000 or so newborns. However, in the Irish traveler population, it affects about 1 in 480.
Huntington Disease (HD)
What is Huntington Disease?
Huntington Disease (HD) is a brain disorder that affects a person's ability to think, talk, and move. People with HD usually experience no effects until they reach age 30, and often even later.
Affected gene
The affected gene in Huntington Disease is the HTT gene. The gene codes for a protein called huntingtin. In people with HD, one of their two copies of the HD gene contains extra copies of the repeated DNA sequence "CAG." People with more copies of this CAG repeat tend to begin having symptoms at a younger age.
The exact job of huntingtin protein is not known. But it is known to be important in nerve cells that send signals in the brain. In people with HD, huntingtin protein contains a long string of the amino acid glutamine. The protein builds up in the brain, possibly sticking together to form clumps. Over time, cells in a part of the brain called the basal ganglia begin to die.
Huntington Disease follows an autosomal dominant inheritance pattern. A child will be affected if they inherit just one disease-causing copy of the gene from one parent. That parent will also be affected.
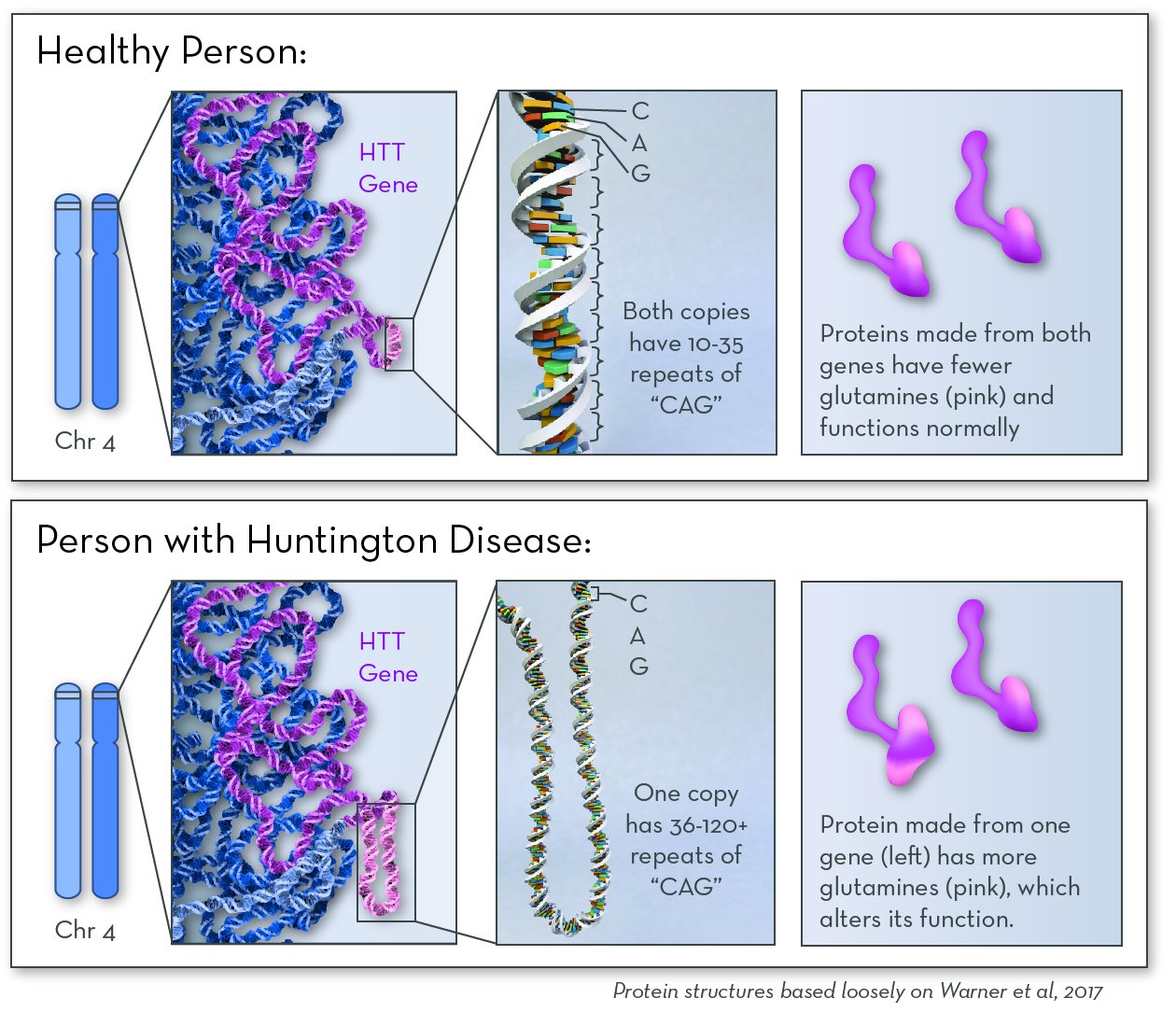
Huntingtin protein is active in neurons in the brain. People with Huntington disease make a version of huntingtin protein that is larger and stickier. It clumps together over time, eventually killing brain cells.
Features and symptoms of Huntington Disease?
Huntington disease affects the part of the brain that controls thinking, emotion, and movement. Most people who have the disease start to see symptoms between the ages of 30 and 50 (but symptoms can appear earlier or later in life). The disease gets worse over time.
Symptoms include poor memory, depression and/or mood swings, lack of coordination, twitching or other uncontrolled movements, and difficulty walking, speaking, and/or swallowing. In the late stages of the disease, a person will need help doing even simple tasks, like getting dressed.
Treating and managing Huntington Disease
Treatments do not slow the progression of the disease, but they can help make the patient more comfortable. Medications ease feelings of depression and anxiety; others control involuntary movements. Physical or speech therapy helps HD patients lead more normal lives.
Interesting facts about Huntington Disease
The disease was named for Dr. George Huntington, who first described it in 1872.
In the United States, about 1 in every 30,000 people has Huntington disease.
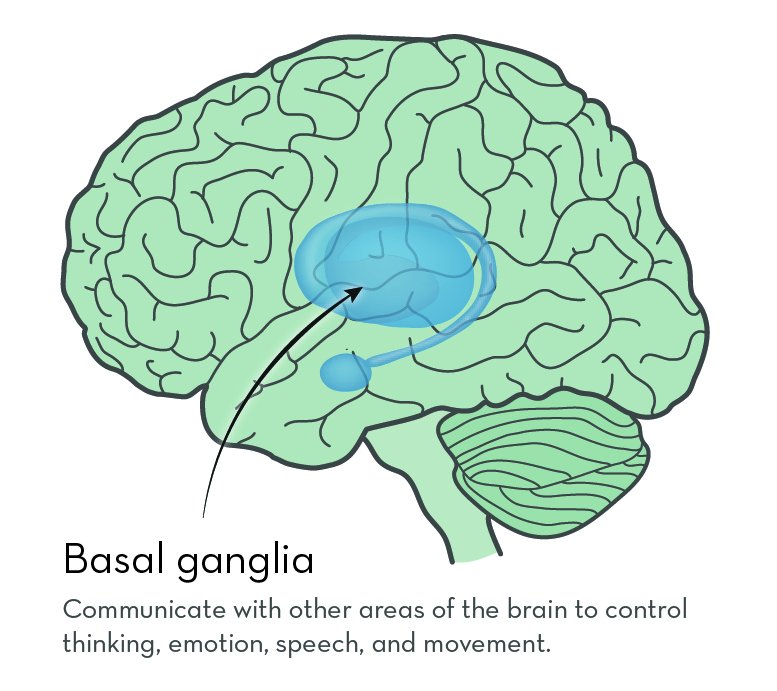
The basal ganglia sit deep inside the brain. They connect to many other brain areas to control thinking, emotion, speech, and movement.
Maple Syrup Urine Disease (MSUD)
What is maple syrup urine disease (MSUD)?
People with MSUD cannot break down three amino acids: leucine, isoleucine, and valine. These amino acids come from protein in food; they are released into the blood stream during digestion. Our cells can either recycle them by adding them to new proteins, or break them down for energy. If they are not used or broken down, they can build up to levels that are harmful to the body.
Affected gene
The most commonly affected gene in MSUD is BCKHDA. MSUD can also be caused by changes in other genes, most commonly BCKHDB and DBT. These genes code for separate proteins that come together as part of a very large complex called BCKD. People with the disorder have a problem with one of the proteins that make up the BCKD complex. Either the protein is missing altogether, or the protein is less able to do its job.
The job of BCKD is to begin breaking down the amino acids leucine, isoleucine, and valine. When any of the protein pieces of BCKD are missing, the whole complex does not work.
MSUD follows an autosomal recessive inheritance pattern. An affected child must inherit two non-working copies of the gene, one from each parent.
To learn more about how proteins from food are broken down into amino acids, visit Digestion.
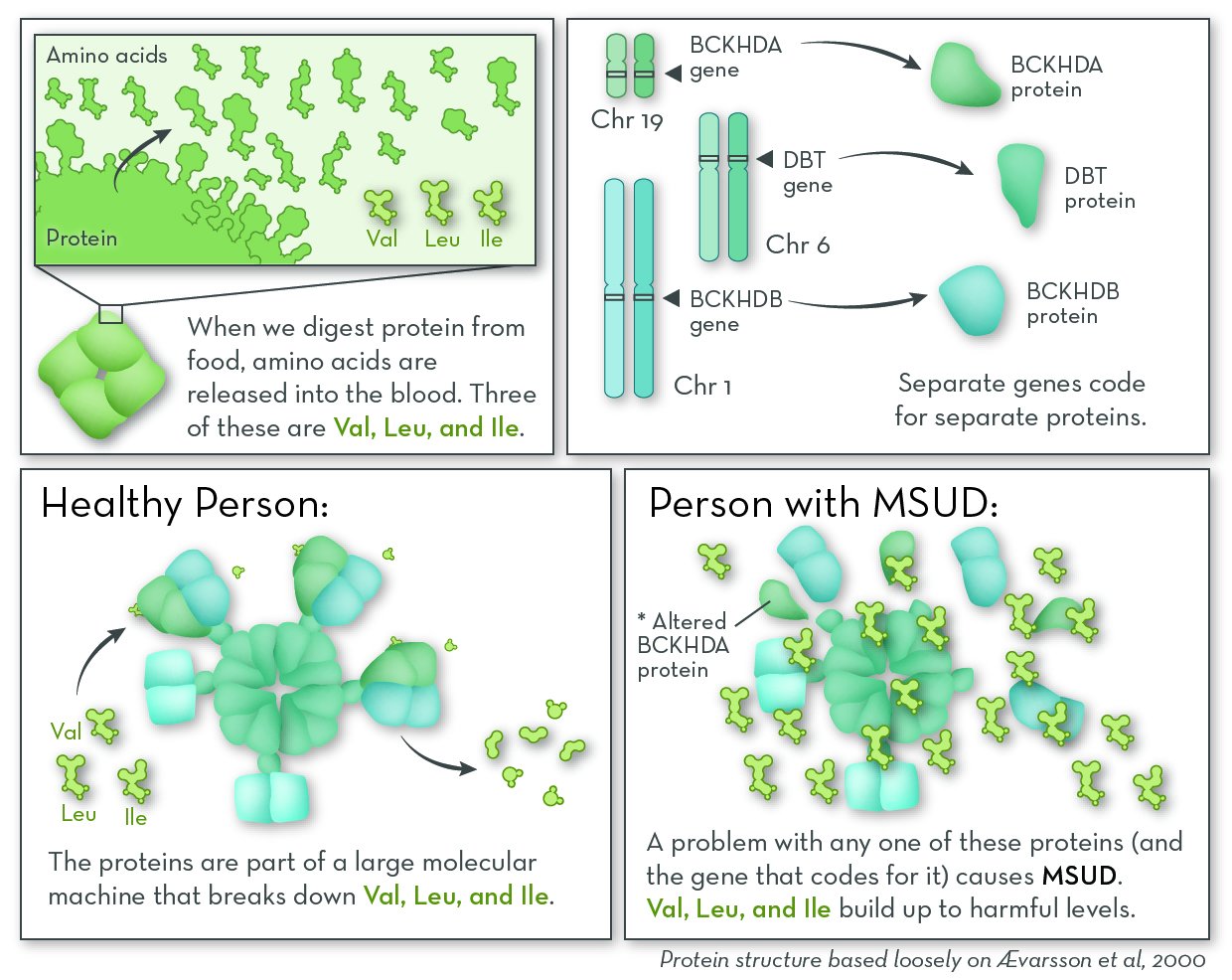
In our cells, a group of genes code for separate proteins that come together and break down the amino acids valine (Val), leucine (Leu), and isoleucine (Ile). A problem with any of these genes, and the proteins they encode, can cause maple syrup urine disease (MSUD).
Symptoms and features of MSUD
MSUD can look different from person to person. The effects depend in part on the specific gene variants that they have. The "classic" form of MSUD is the most common, and also the most dangerous.
Babies who have classic MSUD usually look healthy at birth, but symptoms appear within days. These may include loss of appetite, vomiting, fussiness, and sweet-smelling urine (which smells like maple syrup). Without treatment, babies with the classic form of the disorder usually die within the first few months of life. Other forms, however, have milder symptoms that appear when children are a few weeks, months, or years older.
Because early diagnosis can prevent the most serious symptoms, including brain damage, MSUD is included in most newborn genetic screening panels. Yet even with proper management, many people with MSUD still experience milder effects, such as learning challenges and loss of bone mass (osteoporosis). They are also at risk of having severe symptoms (called a metabolic crisis) when they are sick or stressed, or if they don't eat enough.
Treating and managing MSUD
MSUD is managed through diet. Babies are given a special formula with low amounts of the amino acids leucine, isoleucine, and valine. People with MSUD should avoid eating high-protein foods like meat, eggs, and nuts. Blood and urine tests can be used to check the amounts of amino acids in the body.
If a crisis occurs and the amount of the three amino acids gets too high, people with MSUD can be treated at a hospital to help the levels go back down. Usually people are given a solution through an IV to help the body use up the amino acids by building proteins. A dialysis machine can also be used to filter excess amino acids from the blood.
Interesting facts about MSUD
MSUD is an extremely rare disorder; only 1 in 180,000 babies is born with MSUD. But in certain populations, the disease is much more common. Among the Mennonites in Pennsylvania, as many as 1 out of every 176 babies is born with the disorder.
More information
MSUD Family Support GroupNeurofibromatosis Type 1 (NF1)
What is neurofibromatosis type 1 (NF1)?
Affected gene
The affected gene, also called NF1, codes for a protein called neurofibromin. People with NF-1 have an altered form of the gene, and so they make a form of the protein that does not do its job.
Neurofibromin protein is active in nerve tissue. Its job is to control how often these cells divide to make more cells. It works by deactivating a protein called ras. When ras is active, cells divide quickly; when ras is inactive, cells divide slowly. People with NF1 make a form of neurofibromin that deactivates ras poorly or not at all. Cells divide more often than they should, and the extra cells form tumors.
Having just one non-working copy of neurofibromin is enough to cause NF1. That means that people with NF1 have one working copy of the gene, and they do make some neurofibromin protein that does its job. But they do not make enough to be able to fully control cell division.
NF1 follows an autosomal dominant inheritance pattern. About half of the time, people with NF1 inherit a non-working copy of the NF1 gene from an affected parent. The rest of the time, there is no family history of the disease. Rather, a new mutation in the NF1 gene happens either in the child's parents during egg or sperm production, or soon after fertilization. In either case, the person with NF1 can pass the non-working gene, and the condition, to their children. New mutations in NF1 happen fairly often because the gene is large, making it more likely that a mutation will happen.
To learn how the shape of NF1 protein affects its function, visit Test Neurofibromin Activity in a Cell.
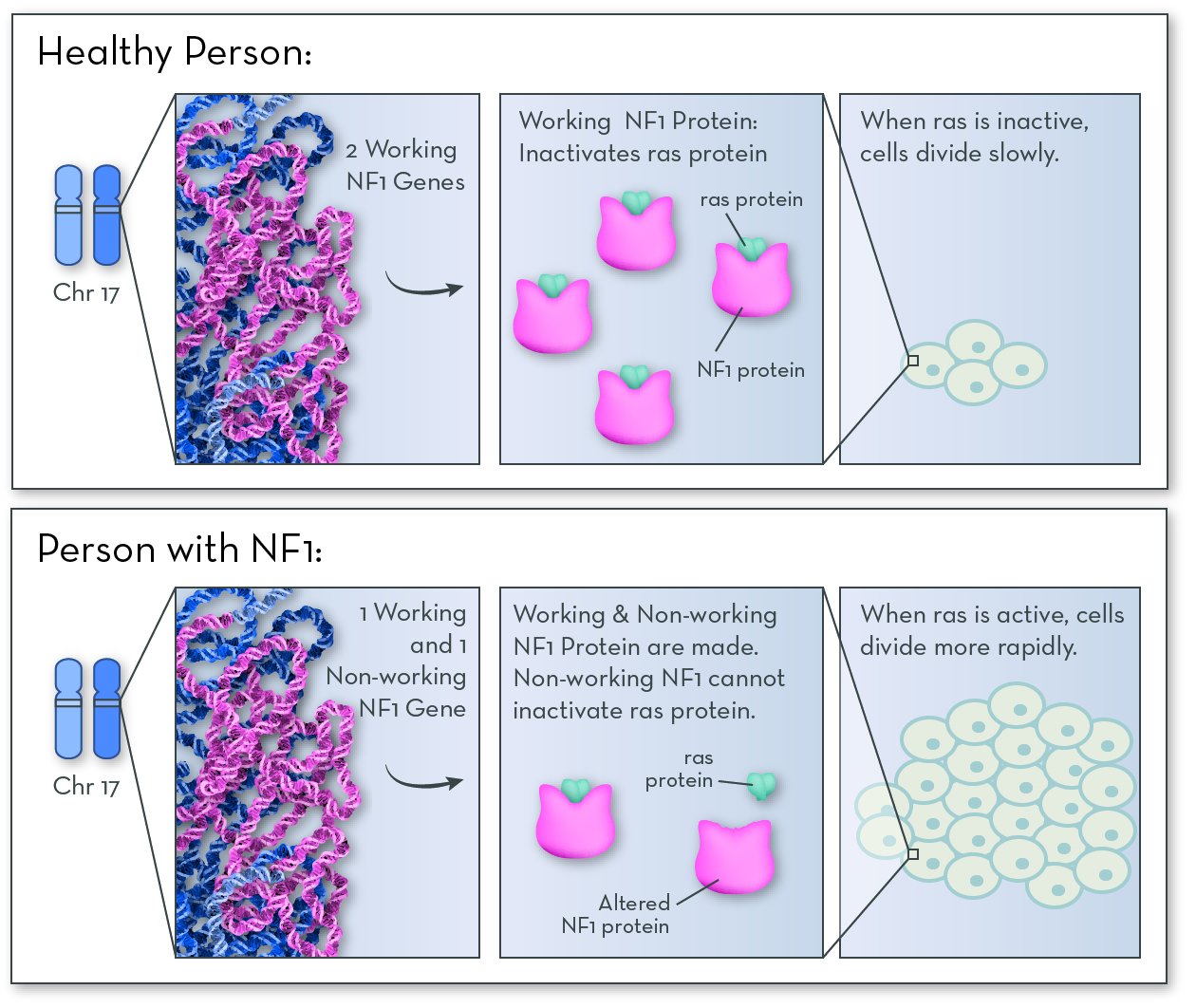
NF1 protein is made in nerve tissue. If NF1 does not work properly, it cannot inactive a protein called ras. Ras causes cells to grow and divide more quickly than normal. The extra cells form bumps and tumors in nerve tissue.
Symptoms and features of NF1?
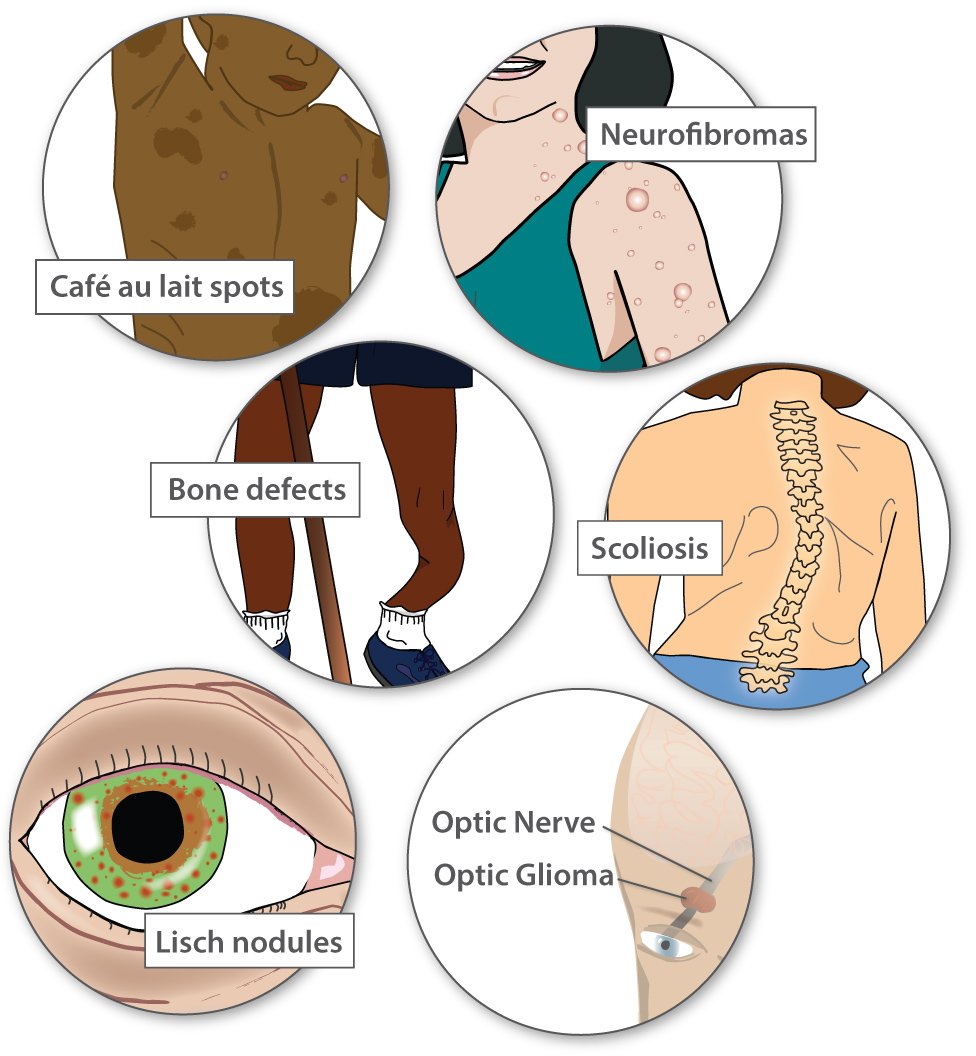
Most people with NF1 have distinctive café au lait spots (the color of coffee with milk), as well as freckles. People get more café au lait spots and freckles as they get older. People with NF1 also have noncancerous tumors called neurofibromas throughout their body. Rarely, these tumors can progress into harmful cancers.
Less common features include high blood pressure, problems with the bones, curvature of the spine (scoliosis), learning challenges, growths on the iris of the eye (Lisch nodules), and non-cancerous tumors on the optic nerve between the eye and the brain (optic gliomas). Some tumors, even when non-cancerous, can cause vision loss or hearing loss.
NF1 can look very different between individuals. The effects depend in part on the specific gene variation that a person has. But they also vary among family members who have the same NF1 gene variation.
Scientists don't know exactly why the effects of NF1 vary so much. But they think that the environment plays a role, as well as other genes (besides NF1). Scientists call these "modifier genes," and they could be any of the thousands of genes in the human genome. Modifier genes may code for proteins that interact directly with neurofibromin or play a role in the same cellular processes.
Treating and managing NF1
Most of the time, NF1 does not have life-threatening medical effects. Surgery can re-shape bones and remove tumors. Bumps and spots can be removed to help people feel about their about their appearance or if they interfere with daily functions. And everyone with NF1 should be monitored for cancer, learning disabilities, and other issues.
Interesting facts
The NF1 gene was first identified as the cause of NF1 in 1990. Over 1,600* different NF1-causing versions of this gene have been found.
NF1 affects about 1 out of every 3,000 people.
Neurofibromatosis was first described in medical literature by Dr. Friedrich von Recklinghausen. For many years, it was called Von Recklinghausen's disease.
* According to the Human Gene Mutation Database, hgmd.cf.ac.uk
Pachyonychia Congenita
What is Pachyonychia Congenita?
Pachyonychia Congenita (PC) is a rare genetic disorder that primarily affects the skin, nails, and mouth. It is caused by a mutation in any one of four genes that code for keratin proteins. Keratins are proteins that form tough fibers that strengthen skin and things that grow out of the skin, such as hair and finger nails. Although mutations in different keratins can cause many disorders, only mutations in keratins 6a, 6b, 16, and 17 are linked to PC. This disorder does not affect lifespan, but patients do experience constant pain.
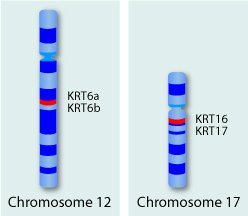
Keratins are a family of related proteins with about 54 members. The keratin genes are located in two clusters, one cluster on chromosome 12 and another on chromosome 17.
How do People Get PC?
PC has a predictable pattern of inheritance. People with PC have a 1 in 2 chance of passing the disease to their children each time they conceive. In other words, the disorder exhibits an autosomal dominant inheritance pattern. Children have to inherit just one copy of the mutated keratin gene to exhibit symptoms.
In some cases, a person with PC will have no family history of the disease. This most commonly happens when a mutation occurs very early in embryonic development, soon after fertilization. It can also occur when a healthy parent's egg or sperm cells acquire a mutation in one of the keratin genes. The mutated gene is passed to the child, resulting in disease. In most cases, the affected child then has a 50% chance of passing PC to each of his or her children.
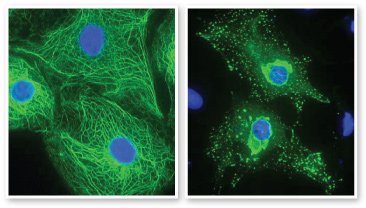
Normal keratin filaments form a strong structural network (left) that enables cells to withstand pressure and stretching. Mutant keratin proteins form clumps (right), resulting in weak cells that break open under pressure. Images courtesy Prof. W.H. Irwin McLean, University of Dundee, Scotland. Keratin filaments are green, and cell nuclei are blue.
What are the Symptoms of PC?
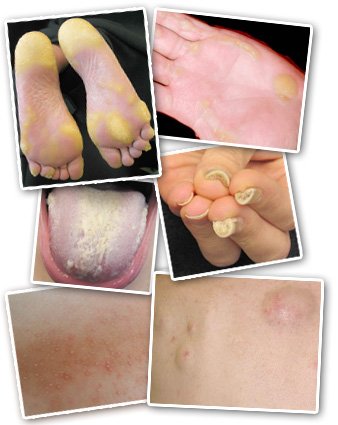
People with PC show a variety of symptoms, including (clockwise from top left) blisters and calluses on the feet and hands, thickened nails, cysts, bumps around hair follicles, and a thick white growth on the tongue.
While every cell in the body has the genes that are involved in PC, symptoms only appear in places where the genes are active. Thick nails are the hallmark symptom of PC. In fact, pachyonychia means "thick nails" However, the most pronounced symptom for people with PC is painful blisters and thick calluses on the soles of the feet. Without proper keratin filaments, skin cells become fragile and cannot withstand pressure or stretching. Just walking across a room can put enough pressure on the soles of the feet to burst skin cells.
Other common PC symptoms include blisters and calluses on the palms of the hands, a white growth on the tongue, and a variety of cysts. Children often have different symptoms than adults. Symptoms more common in children include bumps around hair follicles, hoarseness, and intense, short-duration ear pain. Importantly, individual symptoms and their severity can vary between patients, even within a family.
How do Doctors Diagnose PC?
There are two forms of PC, type 1 and type 2. They are distinguished by slightly different symptoms and by which keratin gene is mutated. To make a diagnosis, doctors typically look for a combination of PC symptoms. Because symptoms can vary, diagnosis is usually confirmed by genetic testing. PC is often misdiagnosed. The most common misdiagnosis for PC is onychomycosis, a fungal infection of the nails. Other common misdiagnoses are epidermolysis bullosa and palmoplantar keratodermas, two skin disorders that are similar to PC.
How is PC Treated?
There is no cure for PC, and drug therapies are limited. Patients typically manage their own symptoms. Trimming and filing nails can prevent infections. Hands and feet can be soaked and rubbed to clean off blistered skin. Keeping tender skin moist and cold is often comforting. Special shoes, canes, or crutches can decrease pressure on the feet.
Work is under way to provide better treatments for PC. A new technology involving gene silencing is providing hope for a cure. By inactivating the mutant keratin gene, researchers hope to restore keratin protein assembly and thereby strengthen skin cells.
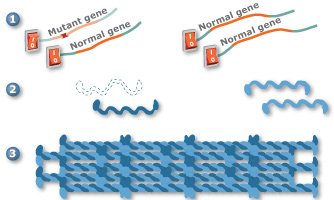
Gene silencing is a promising new technology that may one day be used to treat PC. In one approach to gene silencing, the mutant keratin gene is turned off (1), allowing the three remaining keratin genes to produce proteins (2) that will assemble into proper filaments (3). Scientists are also working on other gene silencing approaches.
Funding provided by the Pachyonychia Congenita Project.
Phenylketonuria (PKU)
What is phenylketonuria (PKU)?
People with PKU cannot break down the amino acid phenylalanine (Phe). Like all amino acids, Phe comes from protein in food. It is released into the blood stream during digestion. Our cells can recycle Phe by adding it to new proteins, break it down for energy, or turn it into other molecules the body needs. When it is not used, Phe can build up to levels that are harmful to the brain.
Affected gene
The most commonly affected gene in PKU is PAH (phenylalanine hydroxylase). The gene codes for the PAH protein. People with PKU have two non-working copies of the PAH gene. Either the protein is missing altogether, or it is less able to do its job.
PAH protein is active in the liver, where it begins the process of breaking down Phe. Without this protein, Phe levels in the blood can become too high.
PKU follows an autosomal recessive inheritance pattern. An affected child must inherit two non-working copies of the gene, one from each parent.
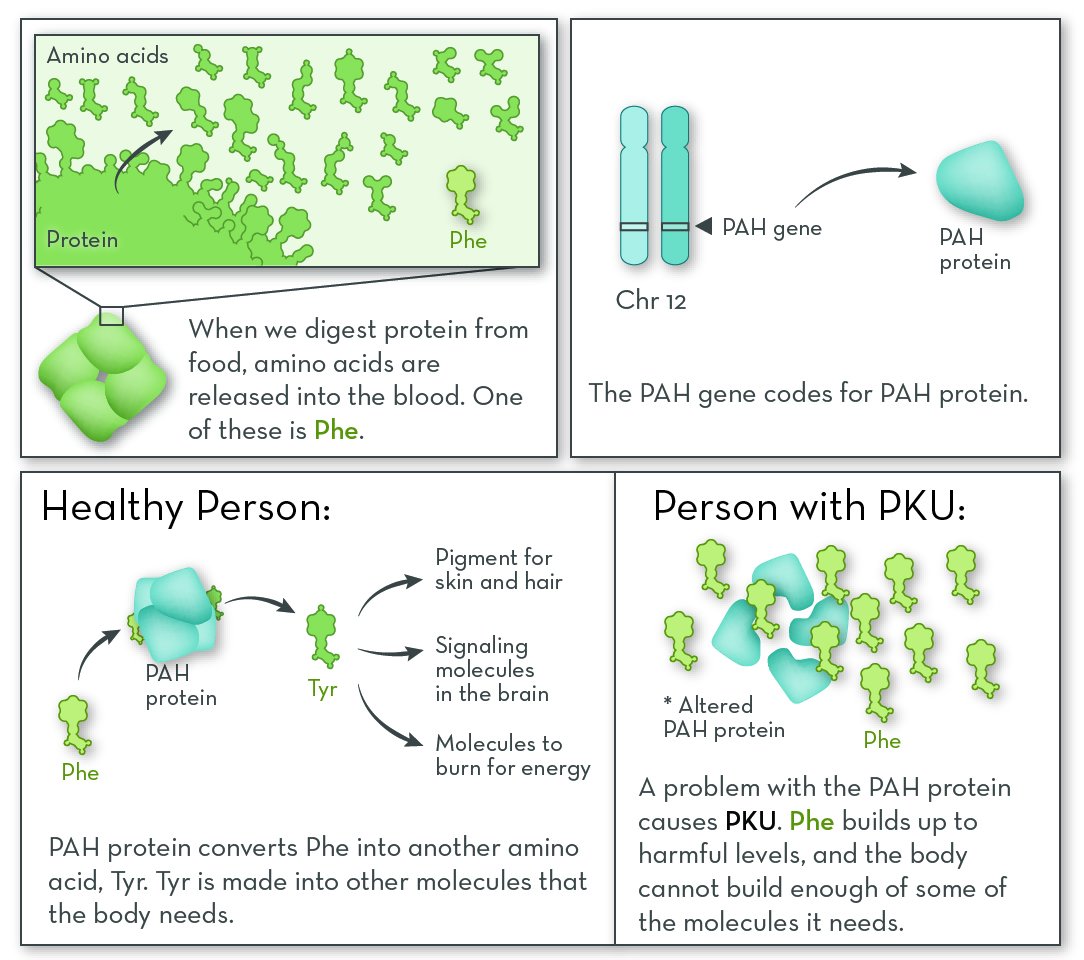
People with PKU are lacking a protein that processes the amino phenylalanine (Phe). They need to eat a diet low in Phe. Because they cannot convert Phe into the amino acid tyrosine (Tyr), they must also take a Tyr supplement.
Symptoms and features of PKU
PKU can look very different between individual people, depending on the specific gene variants they have. The "classic" form is the most common, and also the most dangerous.
Babies who have PKU are usually healthy at first, but symptoms appear within a few months. These may include slow growth, eczema (a skin rash), musty body odor (from too much Phe), small head size, and fair skin (because Phe is converted into skin pigment). Without treatment, children with PKU experience developmental delays, behavior problems, intellectual disability, and seizures.
Because early intervention can prevent the most serious symptoms, PKU is included in most newborn genetic screening panels. With early intervention, many people with PKU have no health effects from their condition.
Treating and managing PKU
PKU is managed mainly with diet, though some people do receive medication to help lower their Phe levels. People who have PKU must eat a very low-protein diet, because nearly all proteins contain Phe. Babies are given a special Phe-free formula. Children must avoid protein-rich foods such as meat, eggs, cheese, and nuts. They must also avoid artificial sweeteners with aspertame, which contains Phe. Adults can usually follow a diet that is less strict.
People with PKU also take a supplement that supplies essential amino acids, including tyrosine (Tyr). These are protein building blocks that the body cannot make on its own and must be gotten through diet.
Interesting facts about PKU
Norwegian doctor Asbjørn Følling discovered PKU in 1934.
About 1 out of every 15,000 babies in the United States is born with PKU.
Severe Combined Immunodeficiency
What is Severe Combined Immunodeficiency (SCID)?
SCID is a group of very rare, and potentially fatal, inherited disorders of the immune system. The immune system normally fights off attacks from bacteria and viruses. People with SCID have a problem in their immune system that leaves them open to infections.
Affected gene
There are several types of SCID, and each is influenced by a different gene. One type, caused by a deficiency of ADA, is described above.
The most common form of SCID is SCID-X1. The affected gene in SCID-X1 is IL2RG (interleuken-2 receptor gamma), on the X chromosome. This gene codes for one of the proteins that make up the IL-2 receptor. People who have SCID-X1 have a non-working copy of the gene, and so their IL-2 receptor does not work.
The IL-2 receptor is active in immune cells, where it helps them communicate with one another when they find bacteria or viruses. When IL-2 is activated, the body makes more immune cells so that it can fight off infection. With no working IL-2, too few immune cells are made, and the body is left defenseless.
SCID-X1 follows an X-linked recessive inheritance pattern. The disorder almost exclusively affects boys, who inherit the disease-causing gene variation from their mothers.
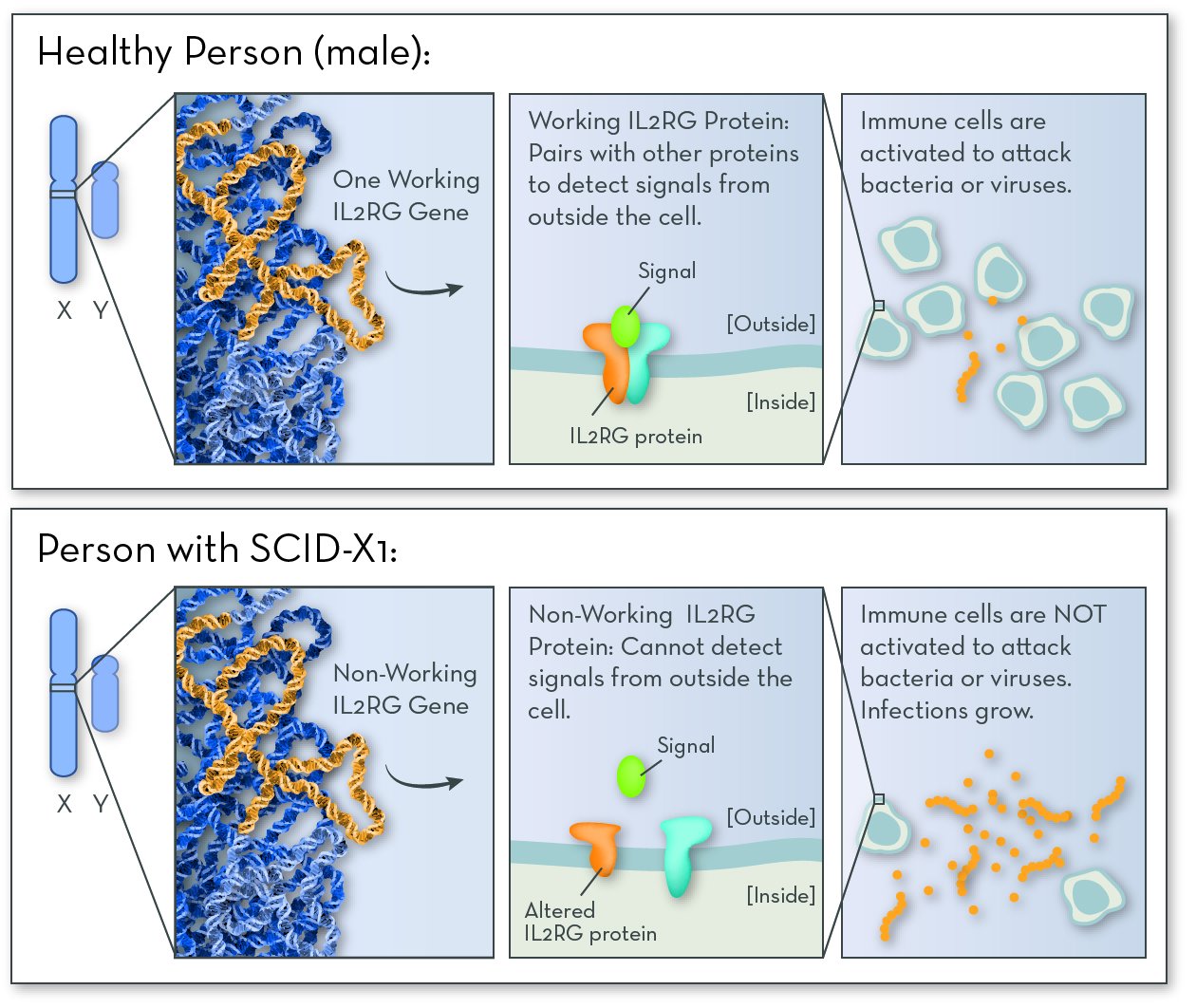
SCID-X1 is caused by a problem with a gene on the X chromosome. Since boys have just one X chromosome, having one non-working copy of the gene causes them to have SCID.
Symptoms and features of SCID
Symptoms usually appear in the first few months of life. Because they have very little immune defense, babies with SCID tend to get one infection after another. They may have a lot of ear infections, sinus infections, a cough that does not go away, and rashes on the skin. Some infections may be life-threatening, including pneumonia (lung infection), meningitis (brain infection), and sepsis (blood infection).
People with SCID often don't respond well to antibiotics, making bacterial infections challenging to treat. With no intervention, children with SCID rarely live beyond age 2.
Interesting facts about SCID
About 1 out of every 100,000 babies is born with SCID.
SCID is sometimes called Bubble Boy disease. In the 1970s, a boy named David Vetter lived in a plastic bubble for 12 years because of SCID.
More information
The Immune Deficiency FoundationTreating and managing SCID
Children with SCID must be careful to stay away from germ-rich environments where they could pick up a life-threatening infection. Most babies who may have SCID are placed into a sterile environment immediately after birth, so that they will not pick up an infection. Antibiotics, anti-viral drugs, and antibodies from the blood of healthy donors can also prevent infection.
One treatment, and potential cure, for SCID is a bone marrow transplant. Bone marrow contains stem cells that give rise to blood cells, including immune cells. Bone marrow stem cells from a healthy donor are injected into the person with SCID. The transplanted stem cells can then give the person with SCID a working immune system.
The biggest challenge to this treatment is finding a donor who is a good genetic "match" for the recipient. Siblings are usually the best donors, as their cells tend to have a more similar genetic makeup. But if parents have one child with SCID, and they have another boy, he will have a 50% chance of also having the condition. Girls will be unaffected, but they may be carriers.
Another way to cure SCID is through gene therapy. Gene therapy places a working copy of the IL2RG gene into the patient's own bone marrow stem cells, and then returns the cells to their body. Gene therapy is relatively new, and there are some technical challenges to getting it to work, but it has been used successfully to treat multiple forms of SCID. It is likely to become less costly and more available over time.
Sickle Cell Disease
What is sickle cell disease?
Sickle cell disease affects a person's red blood cells, which carry oxygen from the lungs to the rest of the body. Normally, red blood cells are round and flexible so they can fit through the narrow blood vessels. But people with sickle cell disease have many long, stiff red blood cells.
Affected gene
The affected gene in sickle cell disease is HBB, on chromosome 11. The HBB gene codes for a protein called beta-globin. People with this disorder still make beta-globin, but the shape of the protein is altered slightly.
Beta-globin combines with another protein, called alpha-globin, to make a larger protein called hemoglobin. Red blood cells are packed full of hemoglobin molecules, which help them carry oxygen to all the tissues of the body.
In people with sickle cell disease, beta-globin proteins stick together to form stiff fibers. Red blood cells become rigid and elongated (like a crescent or a sickle used for cutting wheat). Cells are more likely to become sickle-shaped when oxygen levels are low.
Sickle cell disease follows an autosomal recessive inheritance pattern. An affected child must inherit two non-working copies of the gene, one from each parent.
People who have just one disease-causing gene variant are not only carriers—they also have some resistance to malaria! Malaria is a serious illness that is common in hot climates. It is caused by a parasite that is carried by infected mosquitoes. The parasite cannot live inside of cells with the sticky beta-globin.
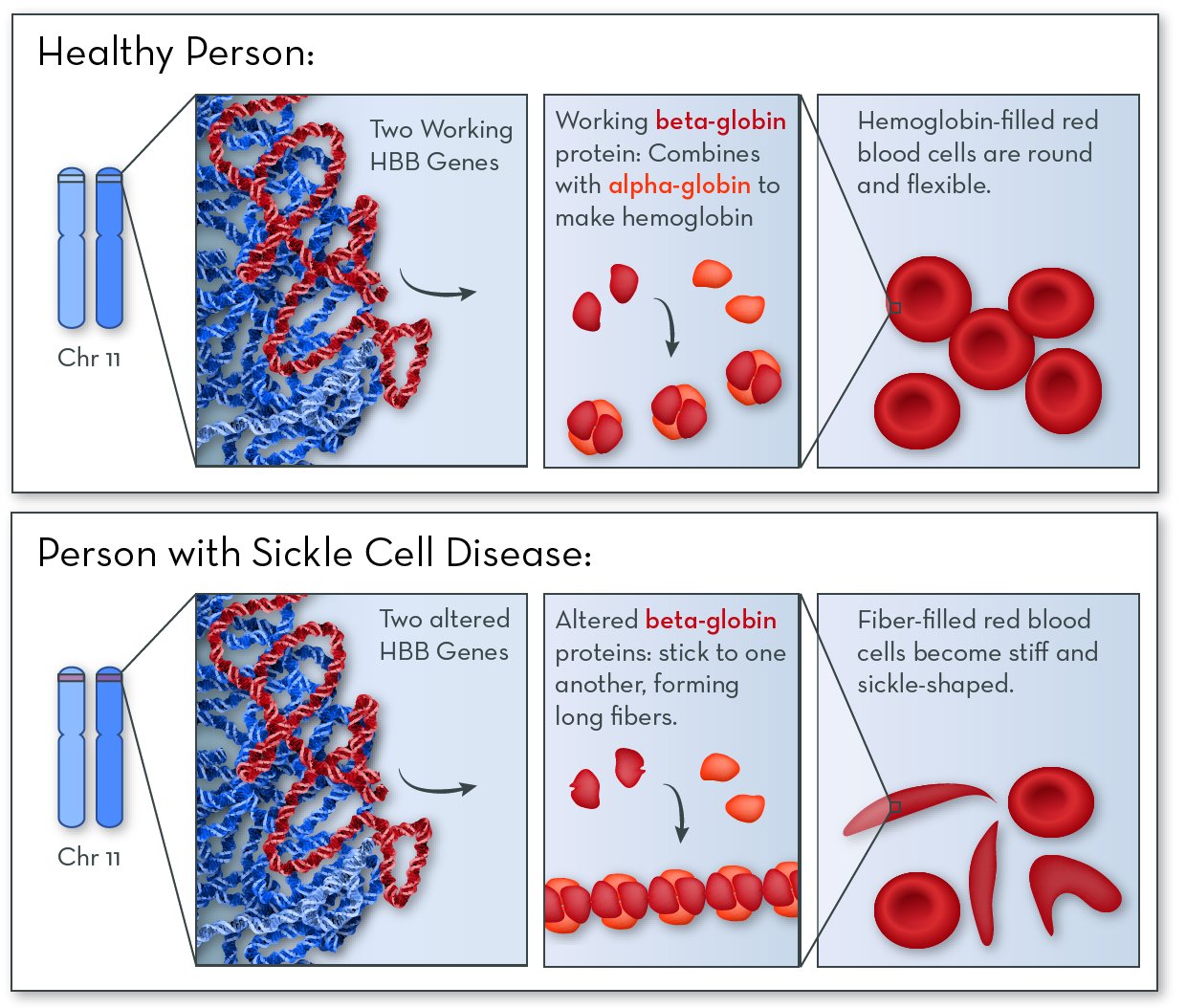
Symptoms and features of sickle cell disease
Symptoms vary between different people who have sickle cell disease, and also in an individual person over time. The symptoms of sickle cell disease come directly from having sickle-shaped red blood cells. The more sickle-shaped red blood cells a person has, the worse the effects.
The stiff, pointy red blood cells can get caught in small blood vessels. This can cause pain and damage organs. The spleen is often affected, along with the liver, kidneys, lungs, and heart. When the spleen is damaged, the body is less able to fight infections.
The sickle-shaped cells are also shorter lived than normal. This can lead to anemia: having too few red blood cells, and less ability to carry oxygen in the blood.
Because early intervention can help to ease symptoms, sickle cell disease is included in most newborn genetic screening panels.
Treating and managing sickle cell disease
For babies and young children, antibiotics can prevent or treat infections. A folic acid supplement can help the body make more red blood cells, helping to prevent anemia.
People with sickle cell disease should get plenty of rest, drink lots of water, and avoid too much physical activity. Physical stress can make it more likely for blood cells to take on a sickled shape. If symptoms become serious, blood transfusions from a healthy donor can provide healthy red blood cells.
In severe cases, people with sickle cell disease can be treated with a bone marrow transplant. Bone marrow contains stem cells that give rise to red blood cells. Bone marrow stem cells are taken from a healthy donor who is a genetic match. The stem cells are then placed into the patient, where can then make a continuous supply of healthy red blood cells.
Interesting facts about sickle cell disease
Red blood cells usually live for 120 days. Sickle-shaped cells live only 10 to 20 days.
In the United States, sickle cell disease most commonly affects African-Americans. In this group, about 1 out of every 500 babies are born with the condition.
Sickle cell disease is most common among people from Africa, India, the Caribbean, the Middle East, and the Mediterranean. These areas also have malaria-carrying mosquitoes. Having some resistance to malaria would give sickle cell carriers in these places a greater chance of surviving childhood and having kids.
Smith-Lemli-Opitz Syndrome (SLOS)
What is Smith-Lemli-Opitz syndrome (SLOS)?
SLOS affects a person's ability to make cholesterol. Cholesterol is required for healthy growth and development. Some cholesterol comes from food, but most of it is made in the body.
Affected gene
The affected gene in SLOS is DHCR7, on chromosome 11. The gene codes for a protein called DHCR7 (7-dehydrocholesterol reductase). People with the disorder have two copies of the gene that have either reduced or no function. They may make very little or no working DHCR7 protein, or they may make a version of the DHCR7 protein with very little activity.
The DHCR7 protein is active in most cells in the body. Its job is to help to make cholesterol. Cholesterol is a fat-like molecule that sits in the membranes of all of the body's cells. It helps membranes stay fluid and flexible. Cholesterol is also converted into other molecules that the body needs, including vitamin D, hormones, and bile salts (which help to digest fats).
With no or no working DHCR7 protein, people with SLOS make very little or no cholesterol. They also have high levels of byproducts of cholesterol metabolism, which can be toxic to the body.
SLOS follows an autosomal recessive inheritance pattern. An affected child must inherit two non-working copies of the gene, one from each parent.
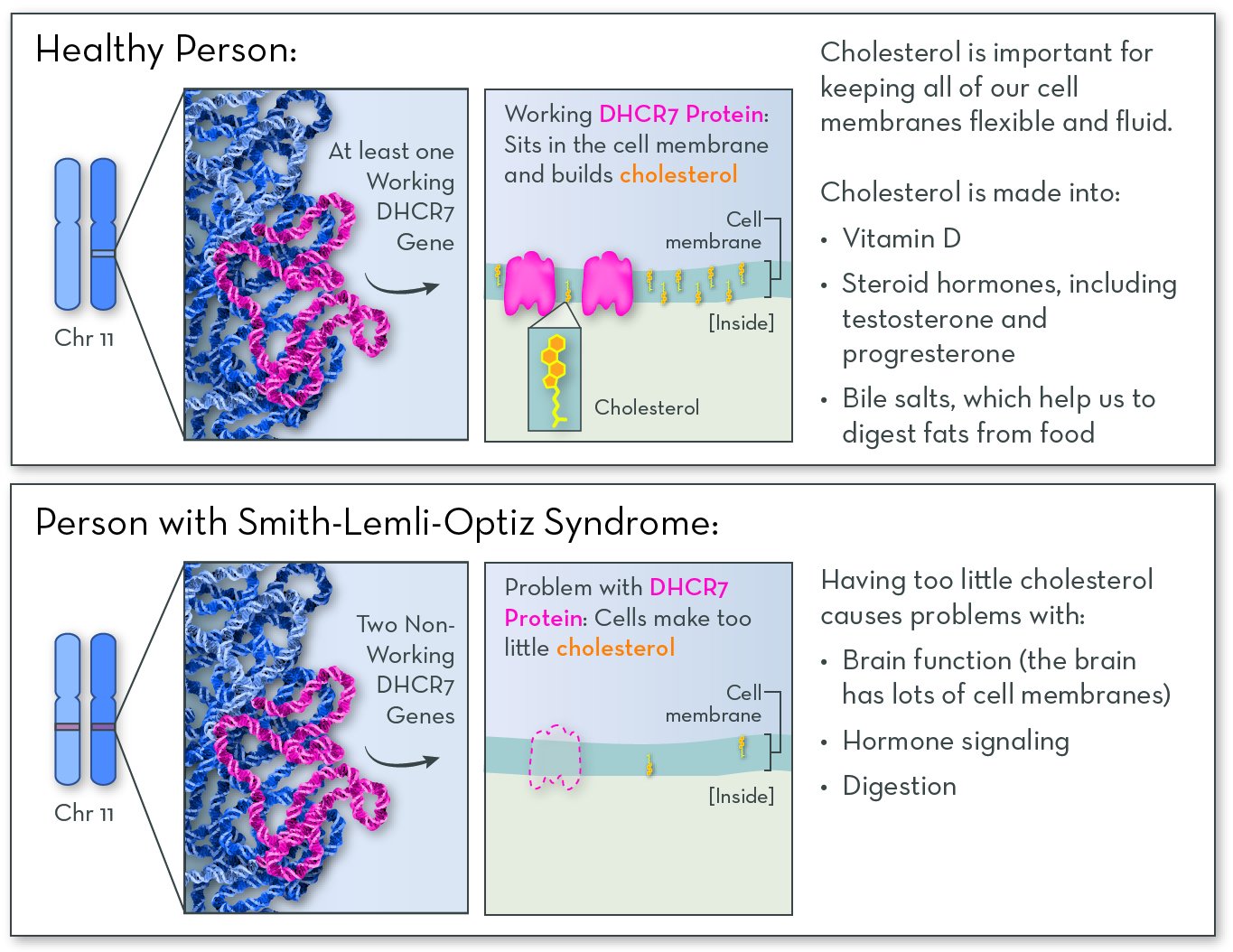
Symptoms and features of SLOS
SLOS can look very different between individual people. The effects depend on the specific gene variants they have, and how much cholesterol they are able to make. Because cholesterol is present in all of the body's cell, SLOS affects many body systems. Cholesterol is important for embryonic development, so many of the characteristics of SLOS form before birth.
The most serious effects of SLOS are on the brain. The brain has a lot of cell membranes, and so it is greatly affected by having too little cholesterol. Children with SLOS often experience small head size, developmental delays, mild to severe intellectual disability, and the characteristics of autism.
Other common features of people with SLOS include distinctive facial features, low muscle tone, low birth weight, slow growth, fused toes (syndactyly), extra fingers or toes (polydactyly), digestive problems, cleft palate (split upper lip), incompletely formed genitals (in males), and sensitivity to sunlight. Less common effects include heart, kidney, and liver problems, hearing loss, and clouding of the eye lens (cataracts).
Treating and managing SLOS
Management for SLOS is based on each person's individual needs. Supplements and medication can supply some of the molecules that people with SLOS cannot make themselves. Babies and adults are usually given cholesterol, bile salts, vitamin D, and certain hormones.
Surgery can repair cleft palate and relieve some of the other physical effects. Clothing and sunscreen protect sensitive skin from sunlight. And physical, occupational, and speech therapies can be a great benefit to young children
Interesting facts about SLOS
In the United States, SLOS affects 1 in 20,000 to 40,000 babies.
SLOS was initially named RSH, for the initials of the first three patients diagnosed with the disorder. It was later changed to honor the three geneticists who first described the disorder in 1964: David Smith, Luc Lemli, and John Opitz.

References
Timson, D. J. (2016). The molecular basis of galactosemia—Past, present and future. Gene, 589(2), 133-141.Huntington disease
Warner IV, J. B., Ruff, K. M., Tan, P. S., Lemke, E. A., Pappu, R. V., & Lashuel, H. A. (2017). Monomeric Huntingtin Exon 1 Has Similar Overall Structural Features for Wild-Type and Pathological Polyglutamine Lengths. Journal of the American Chemical Society, 139(41), 14456-14469.MSUD
Ævarsson, A., Chuang, J. L., Wynn, R. M., Turley, S., Chuang, D. T., & Hol, W. G. (2000). Crystal structure of human branched-chain α-ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Structure, 8(3), 277-291.NF1
Welti, S., Kühn, S., D'angelo, I., Brügger, B., Kaufmann, D., & Scheffzek, K. (2011). Structural and biochemical consequences of NF1 associated nontruncating mutations in the Sec14‐PH module of neurofibromin. Human mutation, 32(2), 191-197.
References
GalactosemiaTimson, D. J. (2016). The molecular basis of galactosemia—Past, present and future. Gene, 589(2), 133-141.Huntington disease
Warner IV, J. B., Ruff, K. M., Tan, P. S., Lemke, E. A., Pappu, R. V., & Lashuel, H. A. (2017). Monomeric Huntingtin Exon 1 Has Similar Overall Structural Features for Wild-Type and Pathological Polyglutamine Lengths. Journal of the American Chemical Society, 139(41), 14456-14469.MSUD
Ævarsson, A., Chuang, J. L., Wynn, R. M., Turley, S., Chuang, D. T., & Hol, W. G. (2000). Crystal structure of human branched-chain α-ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Structure, 8(3), 277-291.NF1
Welti, S., Kühn, S., D'angelo, I., Brügger, B., Kaufmann, D., & Scheffzek, K. (2011). Structural and biochemical consequences of NF1 associated nontruncating mutations in the Sec14‐PH module of neurofibromin. Human mutation, 32(2), 191-197.