Cri-du-Chat Syndrome (chromosome 5 deletion)
What is Cri-du-Chat Syndrome?
Cri-du-chat is French for "cry of the cat," referring to the distinctive cry of children with this condition. The cry is caused by the way the larynx (voice box) develops, one of the features associated with this disorder. It usually becomes less noticeable as the baby gets older, making it harder for doctors to identify Cri-du-chat after age two.
People with Cri-du-chat syndrome have a deletion in one of their copies of chromosome 5. The size of the deletion can vary.
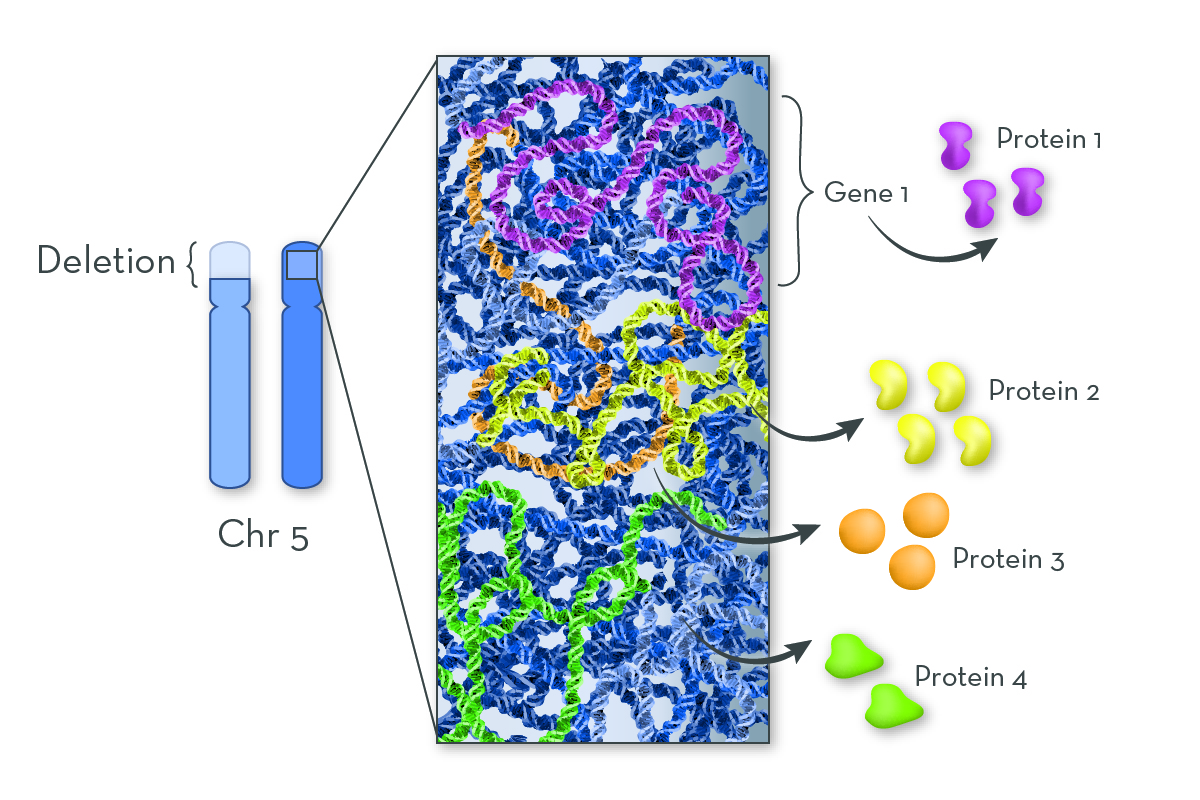
The deleted region in Cri-du-Chat syndrome has many genes—on the order of several dozen to hundreds. For some of these genes, because there is just one copy of the gene instead of two, people with Cri-du-Chat cannot make enough of the proteins the genes code for. The functions of these proteins contribute to the features of the syndrome.
Features and Symptoms

A child with Cri-du-chat syndrome
The features of Cri-du-chat syndrome can vary. They depend in part on how much of chromosome 5 is deleted. Babies with Cri-du-chat are usually small at birth, and they may have problems with their lungs. Often, the larynx doesn't form correctly, which causes the signature cat-like cry.
People who have Cri-du-chat tend to have distinctive facial features. They may have a small head (microcephaly), a round face, a small chin, widely set eyes, folds of skin over their eyes, and a small bridge of the nose.
There can be effects inside the body, as well. Children can have problems with their heart, muscles, bones, hearing, or vision. Most have low muscle tone. As they grow, people with Cri-du-chat usually have difficulty walking and talking. They may have behavior issues, such as hyperactivity or aggression. Most have severe intellectual disability. If there are no major medical problems, life expectancy is about average.
One of the commonly deleted genes in people with this syndrome is TERT (telomerase reverse transcriptase). This gene codes for a protein that helps to keep the tips of chromosomes (telomeres) intact. Other key genes are important for the formation of the throat, larynx, face, and brain.
Interesting facts about cri-du-chat syndrome
The geneticist Jerome Lejeune identified Cri-du-chat syndrome in 1963. He also discovered the genetic basis of Down syndrome.
Cri-du-chat is one of the most common syndromes caused by a chromosomal deletion. It affects between 1 in 20,000 and 1 in 50,000 babies.
In 80 percent of cases, the chromosome carrying the deletion comes from the father's sperm rather than the mother's egg.
If neither parent carries a balanced translocation, it is highly unlikely that they will have a second child with this or any chromosomal deletion.
Williams syndrome (chromosome 7 deletion)
What is Williams syndrome?
Williams syndrome affects a child's growth, physical appearance, cognitive development, and personality.
People who have Williams syndrome are missing genetic material from one of their copies of chromosome 7. The effects are connected to the specific genes that are missing—about 26 to 28 genes in all. With only one copy of each of these genes instead of two, cells cannot make enough of the products that these genes code for.
Deletions happen at this location on chromosome 7 more often than in other places. This is likely because the DNA sequences on either side of the deleted region are highly repetitive. This makes it harder for the chromosomes to line up evenly during the recombination step when egg and sperm are forming.
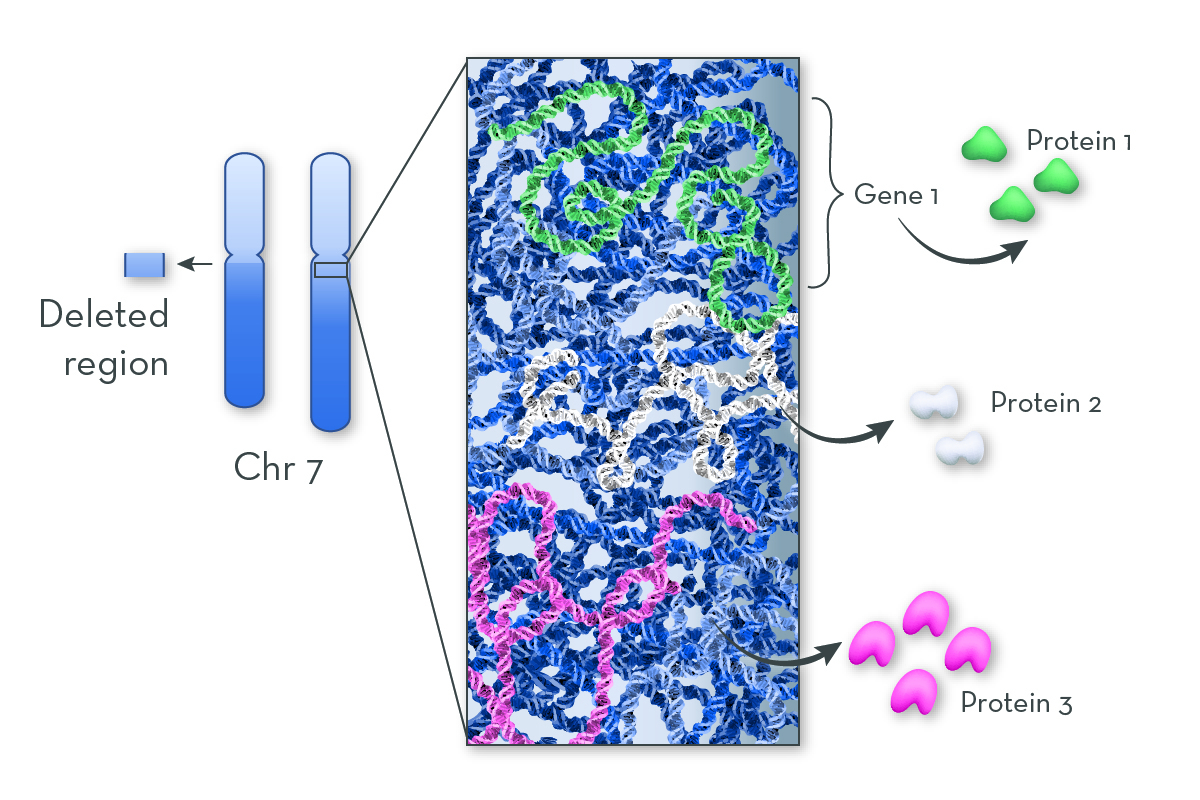
The deleted region in Williams syndrome has many genes. For a key few of these genes, because there is just one copy of the gene instead of two, people with Cri-du-Chat cannot make enough of the proteins the genes code for. The functions of these proteins contribute to the features of the syndrome.
Effects and Features of Williams Syndrome
A child with Williams syndrome
Features vary from person to person. Most people with Williams Syndrome have intellectual disability (usually mild), heart and blood vessel problems, and distinct facial features.
Babies are often born small, and they tend to grow slowly. Children experience developmental delays and low muscle tone. Most have distinct behaviors, such as sensitivity to loud noises and an outgoing personality.
The features of Williams syndrome are connected to the specific genes that are deleted. For example, one of the affected genes is elastin. The elastin gene codes for elastin protein, which helps make blood vessels strong and stretchy. Because they have just one copy of the elastin gene, people who have Williams Syndrome make less elastin protein. This causes problems with the heart and circulatory system.
Diagnosing Williams Syndrome
The deletion in Williams syndrome too small to be seen with a microscope (fewer than 5 million bases of DNA are missing). The small size of the deletion makes Williams syndrome a little trickier to diagnose than other deletion syndromes.
If a person's characteristics strongly suggest Williams syndrome, a doctor may order a test that uses a method called FISH (fluorescent in situ hybridization). If a doctor is less certain about the diagnosis, they may order a test that looks at other chromosomes in addition to chromosome 7. These tests usually use DNA sequencing or microarray analysis.
It is technically possible to diagnose Williams syndrome before birth. But not all prenatal genetic tests look for the specific deletion that causes it.
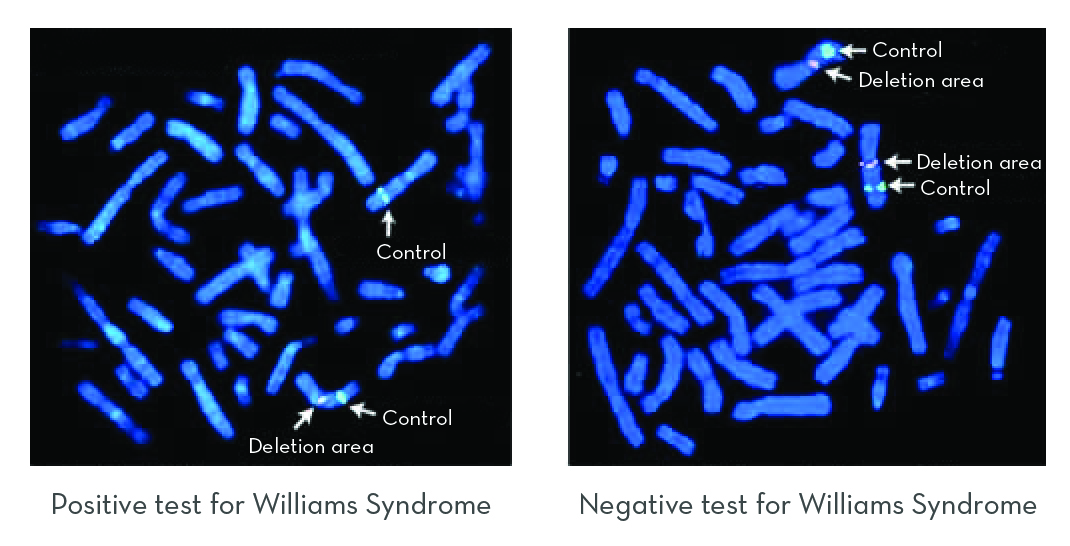
FISH is a method that uses a glowing DNA probe to locate a specific area of a chromosome. Two probes are used to diagnose Williams syndrome. One attaches to the deleted region, and a control probe attaches to a different area of chromosome 7, outside of the deletion.
Interesting Facts About Williams Syndrome
One out of every 10,000 babies is born with Williams syndrome.
If they are genetically normal, the parents of a child with Williams syndrome are unlikely to have a second child with the syndrome.
If someone with Williams syndrome has a child, there is a 50 percent chance the child will also have the syndrome. But most people with Williams syndrome do not have children.
Some cases of Down syndrome arise in the children of parents who carry a Robertsonian translocation that involves chromosome 21. Down syndrome can also be caused by having some duplicated information from chromosome 21, either as an insertion within chromosome 21 or a translocation involving a piece of chromosome 21. Usually the effects of a partial duplication are milder than those of having an entire extra copy of chromosome 21.
Some cases of Turner syndrome are caused by a deletion of a piece of the X chromosome. In these individuals, the effects are usually milder than if they were missing their entire X chromosome.