This page focuses on disorders that affect beta-globin. Similar disorders involve other globins, most commonly alpha-globin.
Hemoglobin Disorders
What are Hemoglobin Disorders?
Hemoglobin disorders are a group of inherited conditions that affect a person's red blood cells. Red blood cells pick up oxygen from the lungs and deliver it to all of the body's tissues. In people with hemoglobin disorders, red blood cells are fewer in number, less able to do their job, or both.
The most common hemoglobin disorders are sickle cell disease and thalassemia. Some version of the genes that cause these diseases also protect against malaria—a deadly parasite carried by mosquitoes. Through natural selection, these gene variations have become very common in certain parts of the world.
Learn what is is like to live with beta-thalassemia.
Affected Gene
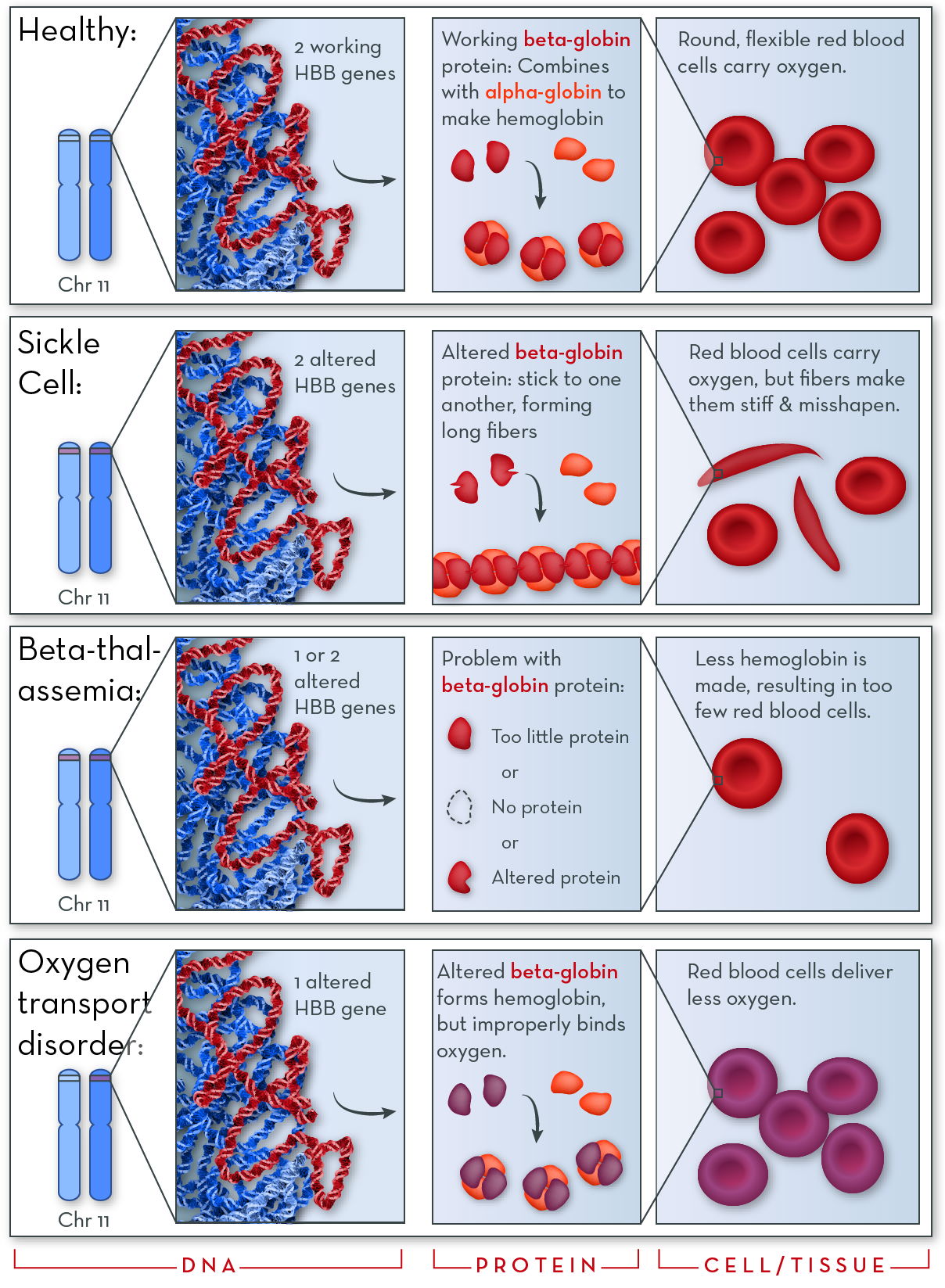
The Hbb gene codes for beta-globin protein. Two molecules of beta-globin combine with two molecules of alpha-globin to form hemoglobin. If there is a problem with beta-globin protein, hemoglobin does not work properly, and red blood cells cannot do their job as well.
The HBB gene, on chromosome 11, codes for beta-globin protein. Two molecules of beta-globin combine with two molecules of alpha-globin to form hemoglobin.
Hemoglobin protein is a major part of red blood cells. It gives blood its color and allows it to carry oxygen. The red comes from hemes—iron-containing molecules that sit within each globin protein. The heme is needed for hemoglobin to hold oxygen.
There are many different versions (alleles) of the HBB gene, each coding for a slightly different beta-globin protein. Some HBB alleles can cause genetic disorders. Each type of beta-globin disorder has a unique set of symptoms, which can range from very mild to life-threatening. In all of these disorders, the symptoms trace back to poorly working hemoglobin, which prevents red blood cells from doing their job.
Problems with beta-globin proteins fall into two broad categories:
Too little protein. Some alleles of the HBB gene produce very little or no beta-globin protein. They cause some forms of beta-thalassemia, a genetic disorder in which people have too few red blood cells.
Altered proteins. Some alleles of the HBB gene code for unusual forms of beta-globin protein. Depending on how the beta-globin protein is altered, alleles of this type can cause multiple genetic disorders.
- In sickle cell disease, modified beta-globin proteins interact differently with each other.
- In some forms of beta-thalassemia, they interact differently with alpha-globin proteins.
- And with oxygen-transport disorders, they interact differently with iron and/or oxygen.
Inheritance
Everyone inherits two copies (alleles) of the HBB gene: one from each parent.
From the perspective of the protein that is made, a person’s two HBB alleles are co-dominant. Beta-globin proteins are made from both alleles, and they combine randomly to make hemoglobin.
Usually, people with one healthy HBB allele make enough healthy beta-globin protein, and their red blood cells can do their job. Thus, hemoglobin disorders usually follow an autosomal recessive inheritance pattern: it takes two non-working alleles to cause the disorder, one from each parent. Sickle cell disease and most forms of beta-thalassemia work this way.
However, in some cases, hemoglobin disorders follow an autosomal dominant inheritance pattern: it takes just one non-working HBB allele to cause the disorder. A child can inherit the disorder directly from an affected parent. Oxygen transport disorders and some forms of beta-thalassemia work this way.
With some allele combinations—like oxygen transport allele plus sickle cell, or sickle cell allele plus beta-thalassemia—the symptoms of the disorder also follow a co-dominant pattern. The symptoms a person experiences are a result of the combined effects of both alleles.
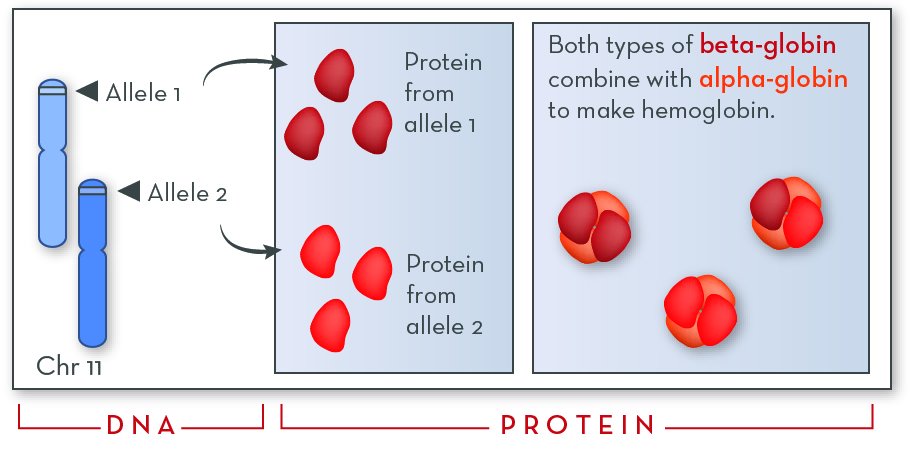
Each person inherits two copies (or alleles) of the Hbb gene—one from each parent. Our red blood cells make protein from both alleles and assemble them into hemoglobin. Hemoglobin molecules can include beta-globin from either allele in any combination.
Normal Protein Expression
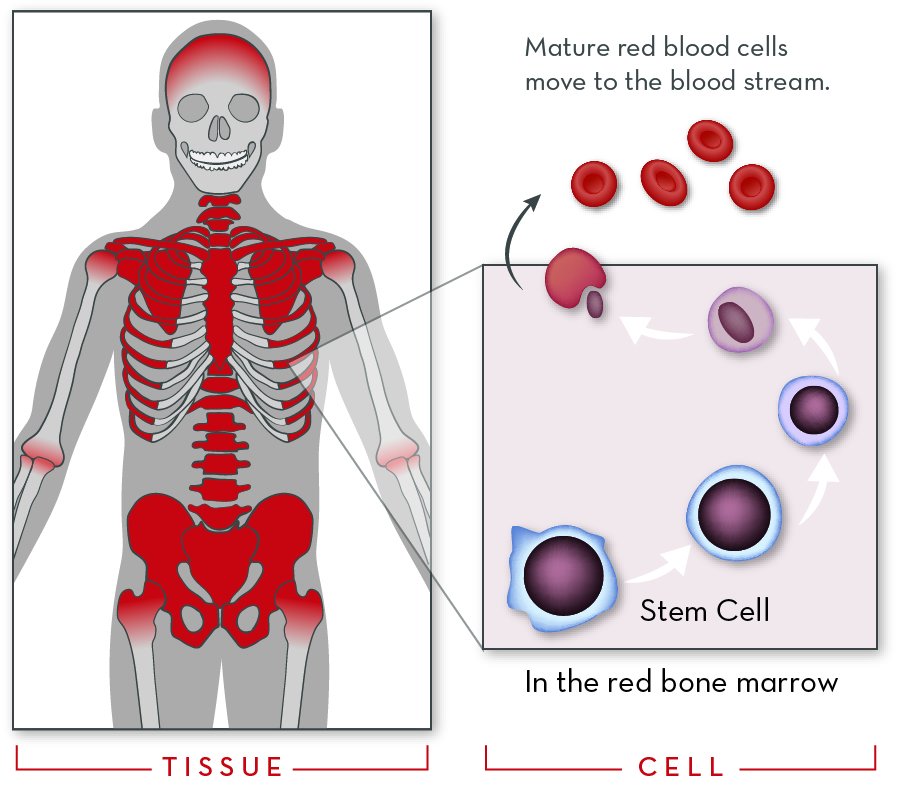
Stem cells in red bone marrow divide rapidly, giving rise to all types of blood cells. The Hbb gene is switched on in ones that will become red blood cells. Once they mature, they move into the blood stream.
Nearly all of the beta-globin in the body is found in red blood cells. A few other cell types make beta-globin protein (and hemoglobin), including cells in the lungs, eyes, and lining of the female reproductive tract. But in these cells, beta-globin is not as central to their function.
Red blood cells are made from stem cells in the bone marrow—more specifically, in red bone marrow. As cells mature, the genes that code for globin proteins are switched on. The cells fill with hemoglobin, the nucleus is pushed out of the cell, and the mature red blood cells enter the blood stream.
Red blood cells live for about 3–4 months, then they are recycled. A healthy adult makes 2–3 million red blood cells every second!
People with beta-globin disorders are born healthy. This is because before we are born, we make a different kind of hemoglobin—called fetal hemoglobin—that uses different globin proteins instead of beta-globin. Shortly before birth, we switch to making beta-globin. These new beta-globin red blood cells gradually replace the fetal hemoglobin ones. Even in the most severe cases, it takes several months for symptoms of a beta-globin disorder to develop.
Protein Function and Interactions
Red blood cells are the largest component of blood tissue, and they are the most plentiful type of cell in the body. They carry out one of blood’s most important jobs: they deliver oxygen to all of the body’s tissues.
Beta-globin protein is essential for the function of red blood cells. It combines with alpha-globin to make hemoglobin—the molecule in red blood cells that carries oxygen. Without healthy beta-globin protein, red blood cells have problems, and blood tissue does not function properly.
Depending on the genetic disorder and the specific HBB alleles involved, people may experience anemia (too few red blood cells), episodes of pain, organ damage, and/or low oxygen levels throughout the body. The next section offers more details.
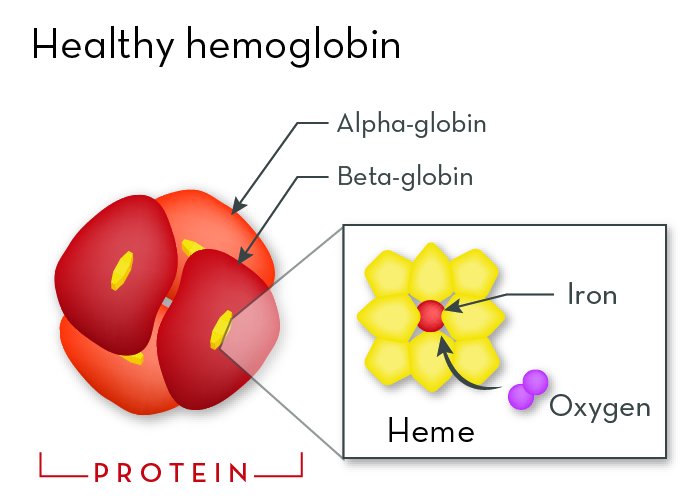
Each globin protein holds an iron-containing heme molecule. The heme helps hemoglobin bind to oxygen. In order for hemoglobin to function properly, beta-globin must interact properly with heme molecules and with alpha-globin.
Symptoms and Features of Hemoglobin Disorders
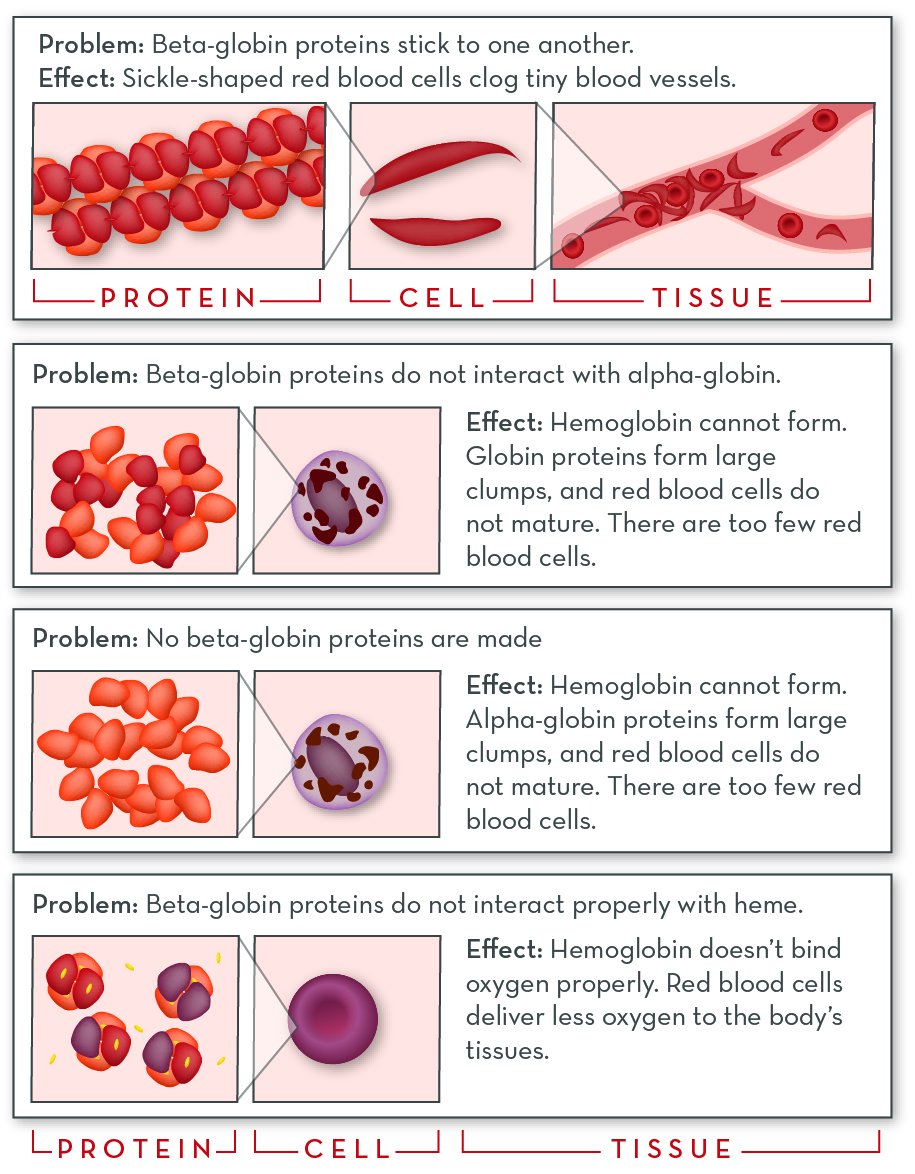
Depending on how beta-globin proteins are affected, hemoglobin disorders can work quite differently. The effects of the disorder are directly tied to the types of beta-globin proteins a person makes.
Hemoglobin disorders are extremely variable from person to person, ranging from mild to life-threatening. And symptoms can appear any time from infancy through adulthood. Though people with different HBB alleles can have very different symptoms, each disorder has defining features. Additionally, people can inherit combinations of HBB alleles that cause disorders with unusual sets of symptoms.
Beta-Thalassemia
People with beta-thalassemia have too few red blood cells. This causes anemia: pale skin, fatigue, loss of breath, slow growth. All of the body’s tissues get less oxygen.
Anemia makes the body work even harder than usual to make red blood cells. But since these cells are short-lived or do not mature at all, people
with beta-thalassemia must also break down more red blood cells than usual. This can cause jaundice (yellowing of the skin) and stress or damage
to the liver and spleen, which do most of the work to recycle blood cells.
People with severe beta-thalassemia can also have toxic effects from iron overload. To get the necessary resources to build red blood cells, the body absorbs more iron than usual from food. Additional iron can come from blood transfusions, which some people need in order to survive.
Sickle Cell Disease
The defining feature of sickle cell disease is rigid, sickle-shaped red blood cells. These cells get stuck in tiny blood vessels. They block blood flow, starving delicate tissues of oxygen.
Complications include episodes of intense pain, infection, organ damage, and stroke. The most vulnerable tissues are the joints, lungs, kidneys,
spleen, and brain.
Many people with this disorder also have too few red blood cells, along with the symptoms of anemia. Their red blood cells go back and forth between being round (when oxygen levels are high) and sickled (when oxygen levels are low). This shape shifting stresses the cells, giving them a much shorter lifespan—just 7–20 days. Some people with SCD need frequent blood transfusions, which increases their risk of iron overload.
Oxygen Transport Disorders
People with oxygen transport disorders usually make enough red blood cells, though some suffer from mild anemia. The main problem is that blood tissue
delivers less oxygen. People may have blue-ish skin, especially in the lips and fingertips; shortness of breath; and purple or brown colored blood.
Alleles, Protein, and Variability
Hemoglobin disorders can look very different from person to person. In general, a person’s symptoms are directly related to the HBB alleles they have, the structure of the beta-globin proteins the alleles code for, and how those proteins affect red blood cells.
HBB alleles are categorized by the type of condition they cause and how well the resulting protein functions. For example, mild beta-thalassemia alleles code for beta-globin proteins that are partially functional. And severe alleles produce either no protein, or proteins that do not function.
The graph shows the concentration of hemoglobin protein in blood samples from people with different HBB allele combinations (who are not receiving treatment). This test alone cannot diagnose a hemoglobin disorder, but it does show how well blood tissue can carry oxygen.
When hemoglobin levels are low, the blood carries less oxygen. The lower the hemoglobin, the sicker the person tends to be.
When doctors diagnose a hemoglobin disorder, they also look at the shape and size of red blood cells. Many diagnostic tests also look at the types and proportions of globin proteins a person makes. And genetic tests, which are often used to confirm a diagnosis, can identify the specific alleles that a person has.
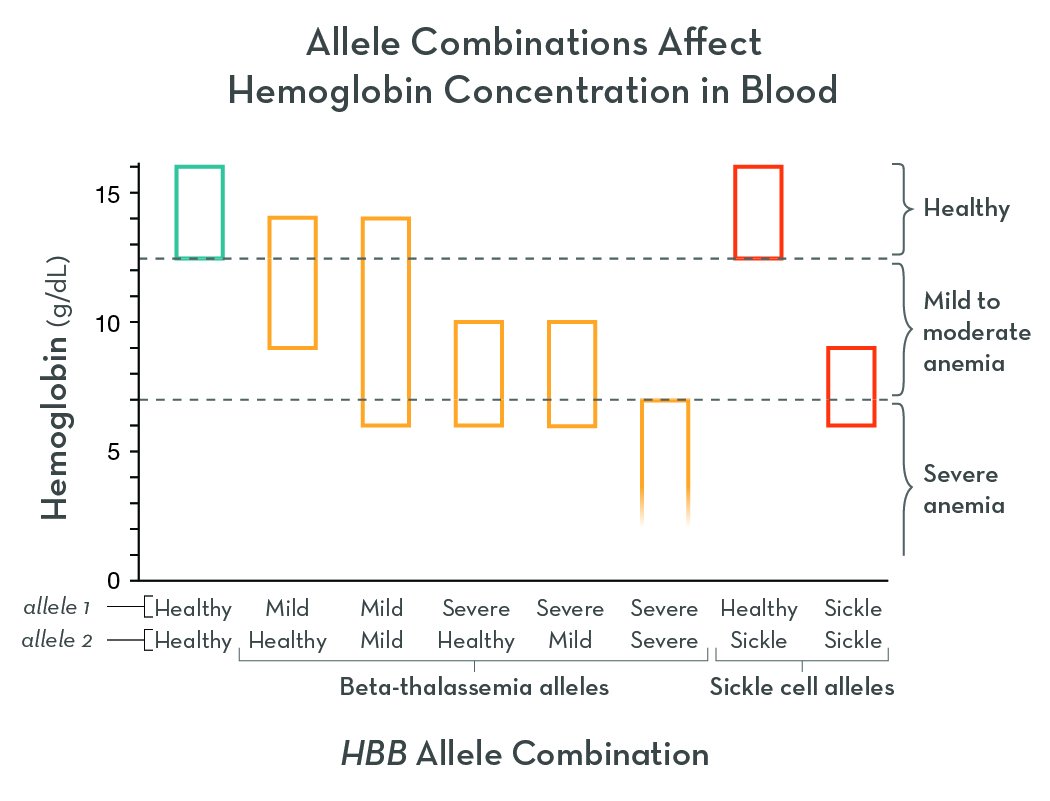
A simple test can measure the amount of hemoglobin in a person’s blood. Anemia is a condition where hemoglobin levels (and red blood cell numbers) are too low. Severe anemia is life threatening. HBB alleles are categorized based on their effects on red blood cell function, and the graph shows the hemoglobin values for each category (in people who are not receiving treatment). Each allele combination has a range of outcomes (Data based on Kohne, 2011).
Other Factors

Even people with the same HBB alleles can have different symptoms.
Even relatives with the same HBB alleles can be affected differently. Some need medical treatment while others remain symptom-free for life.
Some of these differences are due to variations in other genes. For example, genes that code for fetal globins are usually switched off soon after birth. But some people keep making fetal hemoglobin until later in childhood or even into adulthood. As a result, they make more healthy red blood cells, and their symptoms are milder. Other genetic differences affect the amount or type of alpha-globin protein a person makes. This can affect how beta-globin interacts to form hemoglobin, making symptoms better or worse. Still other gene variations affect how well we absorb iron from food. In people with beta-thalassemia, these variations can increase or decrease the risk of iron overload.
Environmental factors also affect the symptoms of hemoglobin disorders. Good nutrition can help the body make and break down red blood cells more quickly. Infections can stress the body and make symptoms worse. Some women experience anemia only during pregnancy, when they need to make more blood to support their growing baby. People tend to do better when they have access to good medical care, and when they closely follow the advice of their caregivers.
Treating and Managing Hemoglobin Disorders
Hemoglobin disorders were some of the first diseases to be understood at a molecular level. Thanks to a series of discoveries beginning in the late 1940s, these once-deadly diseases are now easy to diagnose and treat. Today, people with hemoglobin disorders can use a combination of lifestyle behaviors and medical approaches to manage their health. Yet in many parts of the world, millions of babies and young children each year still die from hemoglobin disorders.
Carriers of certain alleles sometimes have mild symptoms of the disorder. They may develop mild anemia, and those with one sickle cell allele may have some sickle-shaped blood cells when they are at very high altitude, dehydrated, or during extremely rigorous activities. They too should be aware of the lifestyle behaviors listed below.
Lifestyle Behaviors
- Limit alcohol: it stresses the liver.
- Stay well rested and avoid becoming exhausted.
- Those at risk for iron overload should follow a low-iron diet.
- Avoid very high altitude where oxygen levels are lower. With sickle cell diseas, low oxygen makes red blood cells more likely to sickle.
- People with sickle cell disease should avoid vigorous exercise. It lowers oxygen levels, making cells more likely to sickle.
- Stay well hydrated. In sickle cell disease, dehydration causes cells to sickle.
- People with sickle cell disease can use heat and massage during pain episodes.
Medical Approaches
- Vaccinate against influenza, pneumonia, and hepatitis. These preventable illnesses can stress and damage organs. People who receive blood transfusions have a slightly increased risk of hepatitis.
- Blood transfusions provide healthy red blood cells while slowing the productions of ones that will never work properly. People with severe beta-thalessemia may need them every 2-4 weeks, others only during episodes.
- The drug hydroxyurea increases fetal-hemoglobin levels, among other benefits.
- Drugs called chelators can clear excess iron from the body. Monitor iron levels in the blood and organs, and adjust treatment as needed.
- Removing the spleen can relieve pain and improve some symptoms of anemia. In many children with sickle cell disease, the spleen stops working.
- Especially in people with sickle cell disease, antibiotics can treat or prevent infections.
- Vitamin supplements can boost red blood cell production and lower iron levels.
- Anti-inflammatory and pain reliever drugs can help during sickle cell pain episodes.
- If a match can be found, a bone marrow transplant or a cord blood transplant can provide a permanent cure. In both types of transplants, the patient's bone marrow stem cells are replaced by healthy ones from a donor. The donor stem cells make healthy red blood cells.
Beta-globin disorders are a potential candidate for genetic technology. In one experimental approach, a patient’s own cells are modified to correct the problem at the DNA level. Other experimental treatments increase fetal hemoglobin production or decrease alpha-globin production.

Most newborn genetic screening panels include a test for sickle cell disease. People with hemoglobin disorders usually have better outcomes when they are diagnosed at a young age.
More Information
Sickle Cell SocietySickle Cell Information Center
Cooley's Anemia Foundation
Information about beta-thalassemia from the National Organization for Rare Disorders
References
References
Forget, B. G., & Bunn, H. F. (2013). Classification of the disorders of hemoglobin. Cold Spring Harbor perspectives in medicine, 3(2), a011684.
Gerald, P. S., & Efron, M. L. (1961). Chemical studies of several varieties of Hb M. Proceedings of the National Academy of Sciences, 47(11), 1758-1767.
Kohne, E. (2011). Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Deutsches Ärzteblatt International, 108(31-32), 532.
Lohani, N., Bhargava, N., Munshi, A., & Ramalingam, S. (2018). Pharmacological and molecular approaches for the treatment of β‐hemoglobin disorders. Journal of cellular physiology, 233(6), 4563-4577.
Origa, R. (2018). Beta-thalassemia. In GeneReviews®[Internet]. University of Washington, Seattle. Accessed 13 February, 2019.
Patrinos, G. P., Giardine, B., Riemer, C., Miller, W., Chui, D. H., Anagnou, N. P., ... & Hardison, R. C. (2004). Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucleic acids research, 32(suppl_1), D537-D541
Thein, S. L., Hesketh, C., Taylor, P., Temperley, I. J., Hutchinson, R. M., Old, J. M., ... & Weatherall, D. J. (1990). Molecular basis for dominantly inherited inclusion body beta-thalassemia. Proceedings of the National Academy of Sciences, 87(10), 3924-3928.
UniProtKB entry 68871, human HBB. Accessed 13 February 2019 from www.uniprot.org