What is Hemophilia?
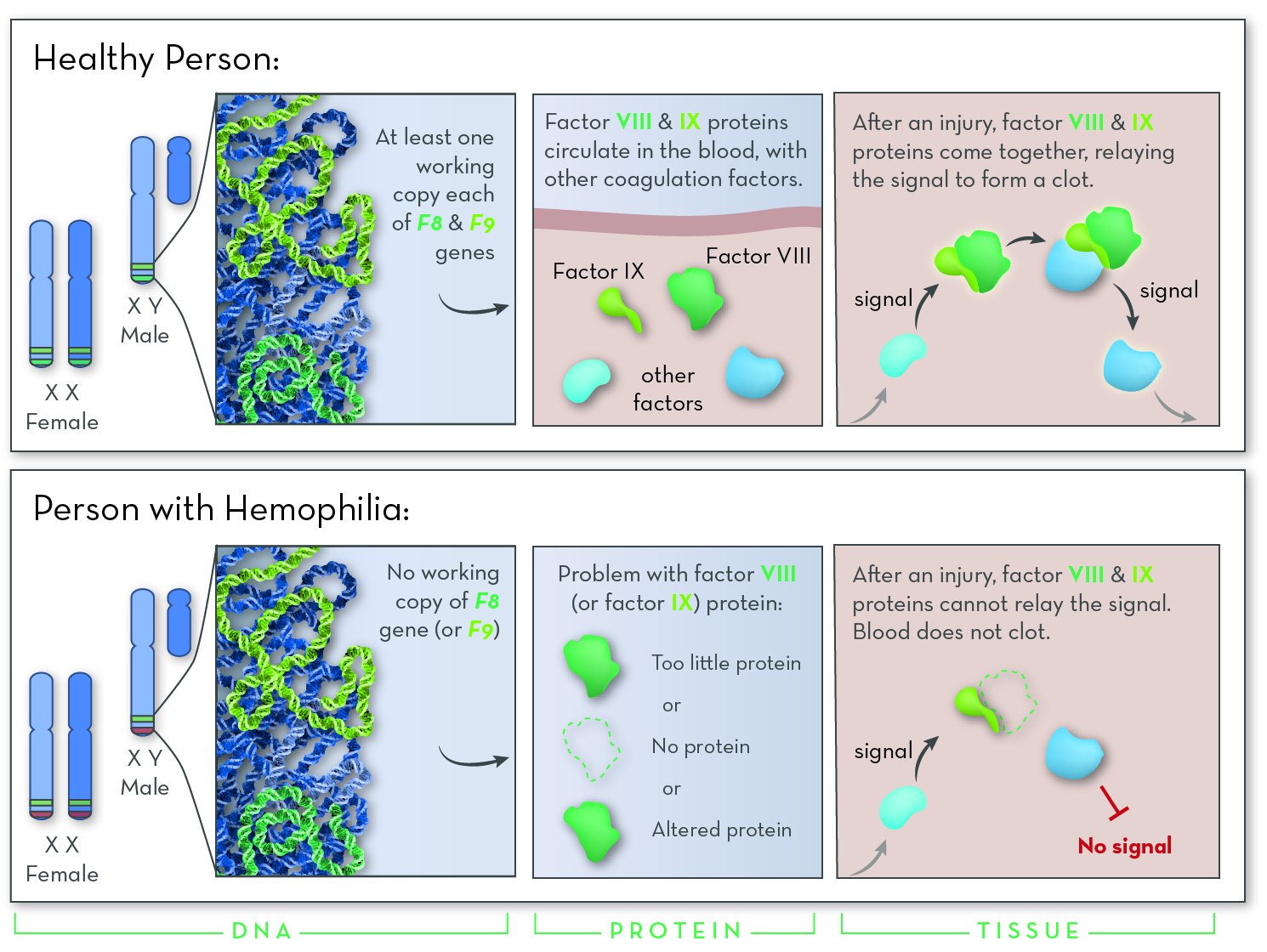
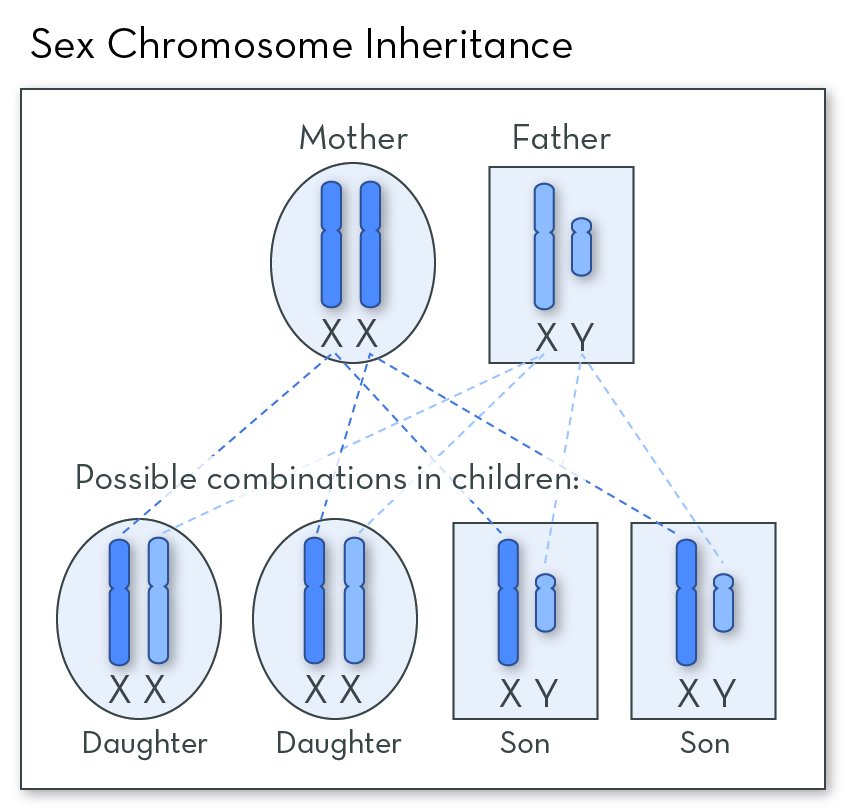
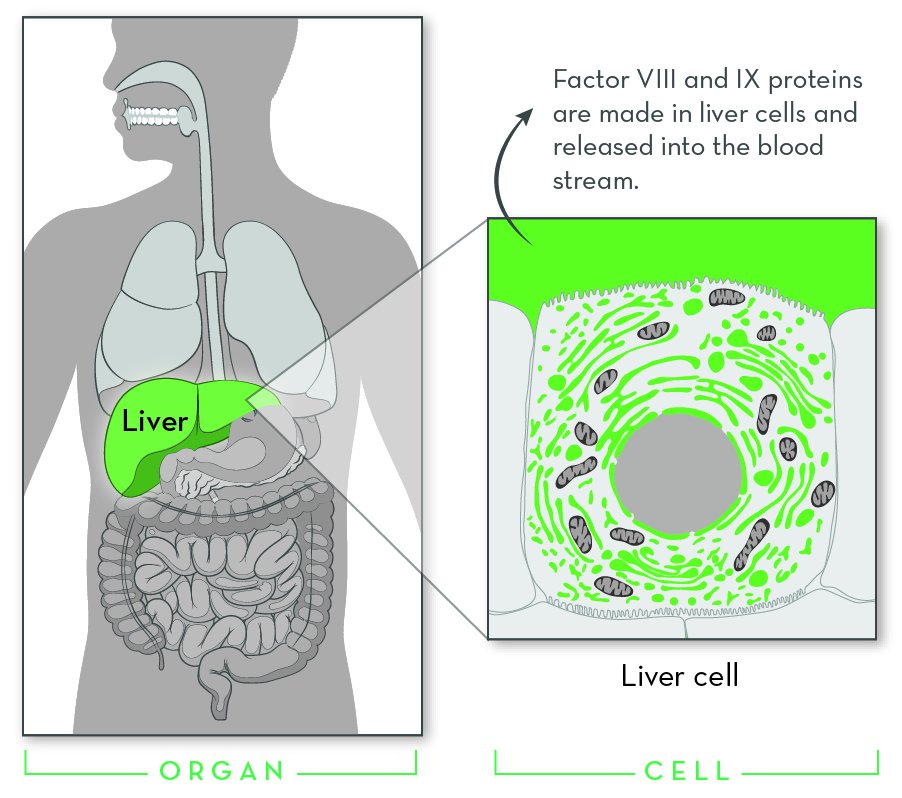
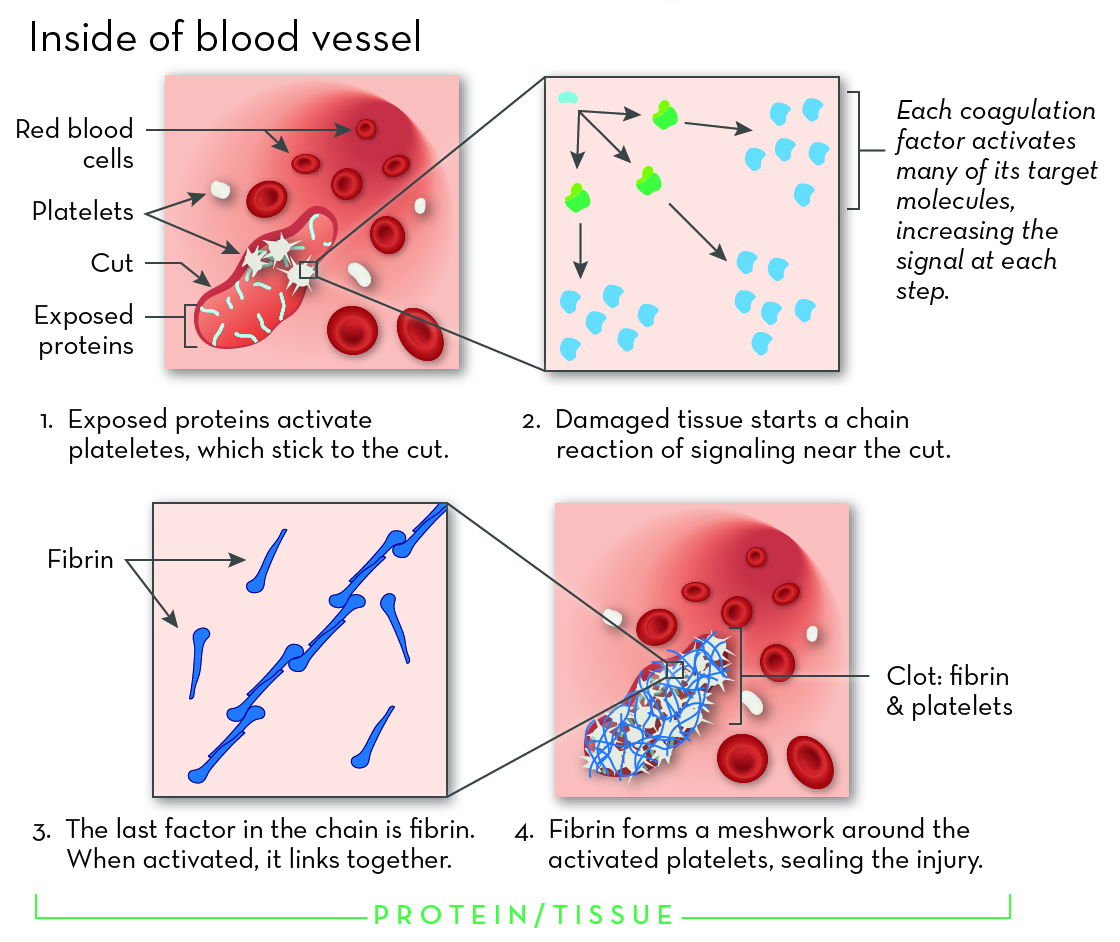
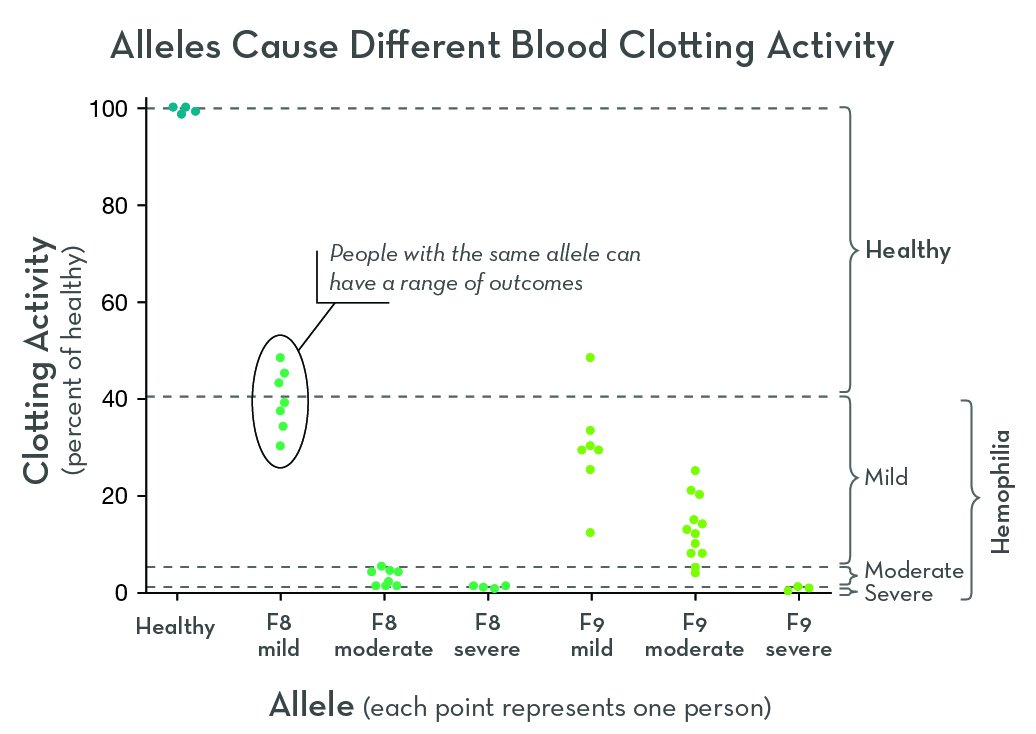
Hemophilia is a genetic disorder that affects blood clotting. The two most common forms are hemophilia A and hemophilia B. Though the cause is different, the main effect is the same: people with hemophilia bleed for longer than normal.
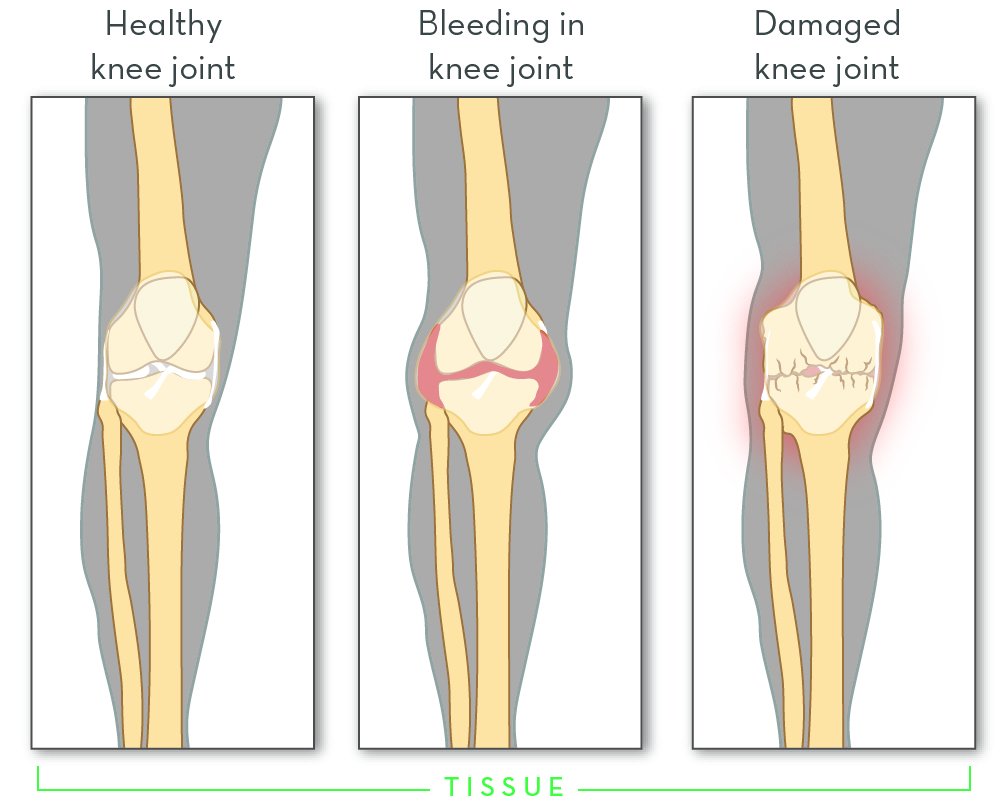
Untreated hemophilia is dangerous. It puts people at risk for spontaneous bleeding that is hard to stop. Bleeding may be internal, for example in the spaces around joints. Repeated bleeding episodes in joints can cause permanent damage.
There is no cure for hemophilia, but there are effective treatments. When hemophilia is detected early and treated properly, the long-term health outcomes are usually very good. Because of this, babies born in families with a history of hemophilia are usually given a genetic test. It is also possible to have hemophilia without a family history. Blood tests are used to diagnose hemophilia in newborns and people who develop bleeding problems later in life.